┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─z╬³ĖĮąį─▄蹊┐
ųąć°╬█╦«╠Ä└Ē╣ż│╠ŠW ĢrķgŻ║2018-7-14 7:40:43
╬█╦«╠Ä└Ē╝╝ąg | ģRŠ█╚½Ū“Łh▒Ż┴”┴┐Ż¼ĮĄĄ═Ų¾śIų╬╬█│╔▒Š
ĪĪĪĪ1 ę²čį(Introduction)
ĪĪĪĪ║¼ųžĮī┘ÅU╦«┼┼Ę┼ĄĮŁhŠ│ųąĢ■įņ│╔ųžĮī┘╬█╚Š, ŲŲē─╔·æBŽĄĮy, ▓ó┐╔═©▀^╩│╬’µ£Ė╗╝»╬Ż║”╚╦ŅÉĄ─ĮĪ┐Ą(Maity et al., 2017).ųžĮī┘╬█╚Šų„ę¬üĒį┤ė┌ĄV╩»ķ_▓╔Īó╗»╣żųŲįņĪóę▒¤ÆĪóļŖÕāĄ╚╣żśI╔·«a▀^│╠.į┌╬ęć°, Ń~╬█╚Šå¢Ņ}ŽÓī”▒╚▌^Ųš▒ķ, ÅU╦«ųąĄ─Cu2+║¼┴┐║▄Ė▀, Ń~╬█╚Šå¢Ņ}žĮąĶĮøØ·Ė▀ą¦Ūę┐╔īŹ¼F┘Yį┤╗ž╩šĄ─ĮŌøQĘĮĘ©(│Ż┤║Ą╚, 2016).─┐Ū░, │Żė├Ą─Cu2+╚ź│²║═╗ž╩š╣ż╦ćų„ę¬ėą╗»īW│┴ĄĒĪóļxūėĮ╗ōQĪó─żĘųļxĪó╬³ĖĮĘ©Ą╚(Hua et al., 2012).Ųõųą, ╬³ĖĮĘ©ę“ŽÓī”ĮøØ·Īó▓┘ū„║å▒ŃĄ╚╠ž³cČ°▒╗ÅVĘ║ĻPūó.─┐Ū░蹊┐║═╩╣ė├▌^ČÓĄ─╬³ĖĮä®ų„ę¬ėą╝{├ū▓─┴Ž(Hua et al., 2012)ĪóĖ▀ÄX═┴(Bhattacharyya et al., 2008)║═╔·╬’┤¾Ęųūė(Triki et al., 2017)Ą╚.
ĪĪĪĪ╦«─²─z╩Ūę╗ĘNį┌╦«ųą─▄čĖ╦┘╚▄├øĄ½▓╗╚▄ĮŌĄ─ĪóŠ▀ėą╚²ŠSĮ╗┬ōŠWĮjĮYśŗĄ─╣”─▄ąįĖ▀Ęųūė▓─┴Ž.ļpŠWĮj╦«─²─z╩Ūė╔ŽÓ╗ź¬Ü┴óŪęŽÓ╗źž×┤®Ą─ā╔éĆ╦«─²─z¾wŽĄśŗ╦∙│╔Ą─, ▌^å╬ŠWĮj╦«─²─zČ°čį, ļpŠWĮj╦«─²─zę“┤¾┤¾╠ßĖ▀┴╦▓─┴ŽĄ─ÖCąĄąį─▄Č°Ą├ĄĮ┴╦▌^ČÓĻPūó(Gong et al., 2003).Ųõųą, ęį╔·╬’Ė▀ĘųūėųŲéõĄ─╦«─²─zę“║¼ėąžSĖ╗Ą─┴u╗∙Īó¶╚╗∙║═░▒╗∙Ą╚╣┘─▄łFČ°Š▀ėą┴╝║├Ą─╬³ĖĮųžĮī┘Ą─ąį─▄.│Żė├Ą─╔·╬’Ė▀Ęųūė╚ńÜżŠ█╠Ū(Haider et al., 2009)Īó├„─z(Wang et al., 2013)Īó└wŠS╦ž(Kono et al., 2013)Īó║ŻįÕ╦ßŌc(Wang et al., 2016)Ą╚, Š▀ėą│╔▒ŠĄ═┴«Īó┐╔╔·╬’ĮĄĮŌąįĄ─ā׳c, ▒╗ÅVĘ║ė├ė┌ŁhŠ│╦«╠Ä└ĒŅIė“.║ŻįÕ╦ßŌc╩Ūę╗ĘN╠ņ╚╗ČÓ╠Ū, ║¼ėą┤¾┴┐Ą─¶╚╗∙║═┴u╗∙, Š▀ėą┴╝║├Ą─│╔─²─ząį─▄, ─▄┼cČ■ār¹}ļxūė(╚ńCa2+ĪóBa2+ĪóCo2+Ą╚)Į╗┬ōČ°─²─z╗».Ą½įōŅÉ─²─zė▓Č°┤Ó, ┐ūŽČ┬╩Ą═, ĘĆČ©ąį▓Ņ(Thakur et al., 2016), į┌ę╗Č©│╠Č╚╔ŽŽ▐ųŲ┴╦Ųõæ¬ė├ĘČć·.Š█ęꎮ┤╝╩Ūę╗ĘN╩╣ė├ÅVĘ║Ūę¤oČŠ¤o║”Ą─╦«╚▄ąįĖ▀ĘųūėŠ█║Ž╬’, ┐╔═©▀^裣h└õā÷ĮŌā÷Ą─ĘĮ╩Įą╬│╔─²─z.┼c║ŻįÕ╦ßŌc─²─zŽÓ▒╚, Š█ęꎮ┤╝─²─zŠ▀ėą┴╝║├Ą─Ēgąį║═ĘĆČ©ąį, Ą½ė▓Č╚▌^Ą═, Ūę╣┘─▄łFå╬ę╗(Yang et al., 2016), į┌ŁhŠ│ĘĮ├µæ¬ė├▌^╔┘.┤╦═Ō, ųTČÓ╝{├ū▓─┴Ž, ╚ń┤┼ąį╝{├ū┴Żūė(Li et al., 2016)Īó╠╝╝{├ū╣▄(Karkeh-abadi et al., 2016)Īó╩»─½Ž®(Li et al., 2013)Ą╚▒╗ė├ė┌┼c╦«─²─zÅ═║Žęį▀Mę╗▓ĮĖ─╔ŲŲõ╬³ĖĮąį─▄ĪóÖCąĄąį─▄║═╔·╬’ĘĆČ©ąį, ▓ó½@Ą├Ė³ĘĮ▒ŃĄ─╣╠ę║Ęųļx─▄┴”.Ųõųą, ┤┼ąį╝{├ūFe3O4┴Żūėę“ųŲéõ╣ż╦ć║åå╬Īó▒╚▒Ē├µĘe▌^Ė▀ĪóŠ▀ėą│¼Ēś┤┼ąį║═▌^║├Ą─╬³ĖĮąį─▄Č°éõ╩▄ĻPūó(Mohammadi et al., 2014), Ą½┤┼ąį╝{├ūFe3O4┴Żūė▒®┬Čį┌┐šÜŌųąęū▒╗č§╗»Īóęū░l╔·łFŠ█ŪęĘĆČ©ąį▓Ņ, ĮĄĄ═┴╦Ųõæ¬ė├ąį─▄.
ĪĪĪĪ×ķ┴╦śŗĮ©ōĒėąžSĖ╗╣┘─▄łFęį─▄ē“Ė▀ą¦╬³ĖĮ╚ź│²ųžĮī┘ļxūėŪęŠ▀ėą┴╝║├╣╠ę║Ęųļxąį─▄Ą─┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─z, ▒ŠčąŠ┐öMį┌║ŻįÕ╦ßŌc─²─z╗∙ĄA╔Ž, ═©▀^┼cŠ█ęꎮ┤╝─²─z║═┤┼ąį╝{├ūFe3O4Ą─Å═┼õ, ęį║ŻįÕ╦ßŌc┼cCa2+Į╗┬ōą╬│╔Ą┌ę╗ŠWĮj, Ą├ĄĮ┤┼ąįå╬ŠWĮj╦«─²─z(║åīæ×ķSAPFe); ┤┼ąįå╬ŠWĮj╦«─²─z└^└m═©▀^Š█ęꎮ┤╝└õā÷ĮŌā÷裣hą╬│╔Ą┌Č■ŠWĮj, ½@Ą├ļpŠWĮj┤┼ąį╦«─²─z(║åīæ×ķDAPFe).═¼ĢrŻ¼└¹ė├Æ▀├ĶļŖńRĪó╝t═Ō╣ŌūVĪó║¼╦«┬╩ūā╗»Īó▒╚▒Ē├µĘeī”SAPFe║═DAPFe▀Mąą▒Ēš„, ▓ó═©▀^ī”Cu2+Ą─╬³ĖĮīŹ“×╠ĮėæSAPFe║═DAPFeĄ─╬³ĖĮąį─▄.
ĪĪĪĪ2 ▓─┴Ž┼cĘĮĘ©(Materials and methods)
2.1 ų„ę¬▓─┴Ž
ĪĪĪĪ║ŻįÕ╦ßŌcĪóŠ█ęꎮ┤╝Īó┬╚╗»Ō}Īó┴“╦ßŃ~Īó┴∙╦«║Ž┬╚╗»ĶFĪóęęČ■░ĘĪóÜõč§╗»ŌcĪóÖÄ├╩╦ßŌcĄ╚╦Ä䮊∙×ķĘų╬÷╝ā, ┘Åūįć°╦Ä╝»łF╗»īWįćä®ėąŽ▐╣½╦Š.īŹ“×ė├╦«Š∙×ķ│¼╝ā╦«.
ĪĪĪĪ2.2 ┤┼ąį╝{├ūFe3O4Ą─ųŲéõ
ĪĪĪĪ╦«¤ßĘ©ųŲéõ┤┼ąį╝{├ūFe3O4Ż║╚Ī0.2 g┴∙╦«║Ž┬╚╗»ĶF╚▄ĮŌų┴15 mL│¼╝ā╦«ųą, Ęųäe╝ė╚ļ0.5 gÖÄ├╩╦ßŌcĪó3 mLęęČ■░ĘĪó0.3 gÜõč§╗»Ōc, öć░ĶŠ∙ä“║¾╝ė╚ļĖ▀£žĘ┤æ¬Ė¬ā╚, ╝ė¤ß200 Īµ▒Ż│ų12 h.«a╬’▀Mąą┤┼Ęųļx║¾ė├ęę┤╝Īó│¼╝ā╦«╦«Ė„ŪÕŽ┤3┤╬, šµ┐š║µĖ╔║¾éõė├.
ĪĪĪĪ2.3 å╬ŠWĮj┤┼ąį╦«─²─z(SAPFe)Ą─ųŲéõ
ĪĪĪĪīó2 g║ŻįÕ╦ßŌc║═2 gŠ█ęꎮ┤╝╚▄ĮŌĄĮ100 mL│¼╝ā╦«ųą, ╚Ī0.1Īó0.2Īó0.5Īó1.0 gųŲéõ║├Ą─┤┼ąį╝{├ūFe3O4╝ė╚ļ╔Ž╩÷╚▄ę║, ÖCąĄöć░Ķ║═│¼┬Ģ½@Ą├Š∙ę╗╚▄ę║, ═©▀^╚õäė▒├Ą╬╚ļ100 mL 10%Ą─CaCl2╚▄ę║└’, Ą├ĄĮžō▌d┤┼ąį╝{├ūFe3O4┴┐×ķ0Īó2.5%Īó5.0%Īó12.5%Īó25.0%Ą──²─zŪ“.Į■┼▌24 h║¾, ė├│¼╝ā╦«ŪÕŽ┤3┤╬, ╚ź│²▒Ē├µĄ─ļs┘|ļxūė, ½@Ą├SAPFe.
ĪĪĪĪ2.4 ļpŠWĮj┤┼ąį╦«─²─z(DAPFe)Ą─ųŲéõ
ĪĪĪĪīó2.3╣ØųŲéõĄ─žō▌d┤┼ąį╝{├ūFe3O4┴┐Ęųäe×ķ0Īó2.5%Īó5.0%Īó12.5%Īó25.0%Ą─å╬ŠWĮj╦«─²─zŪ“čb╚ļ┼ÓB├¾ųą, į┌-40 ĪµŽ┬└õā÷24 h, į┘│Ż£žŽ┬═Ļ╚½ĮŌā÷.╔Ž╩÷▓┘ū„▀Mąą3éĆ裣h, ╦∙Ą├─²─zŪ“ė├│¼╝ā╦«ŪÕŽ┤3┤╬╝┤Ą├ĄĮDAPFe.
ĪĪĪĪ2.5 ┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zĄ─▒Ēš„┼cĘų╬÷ĘĮĘ©
ĪĪĪĪ╦∙ėąśėŲĘČ╝Įø▀^└õā÷Ė╔į’║¾▀Mąą▒Ēš„Ęų╬÷, śėŲĘĄ─▒Ē├µą╬├▓└¹ė├╚š▒Š╚š┴ó╣½╦ŠĄ─ł÷░l╔õÆ▀├ĶļŖūė’@╬óńR(SEM, H-7500)½@Ą├; śėŲĘ┐ūĮYśŗ└¹ė├├└ć°¹£┐╦╣½╦ŠASAP-2460╚½ūįäė▒╚▒Ē├µĘe┼c┐ūŽČČ╚āxĘų╬÷, śėŲĘį┌80 Īµ├ōÜŌ4 h, ė┌ę║Ą¬(77 K)£žČ╚Ž┬ęįĖ▀╝āĄ¬×ķ╬³ĖĮĮķ┘|£yČ©ŽÓī”ē║┴”×ķ0~1Ą─╬³├ōĖĮŪ·ŠĆ; ĖĄ└’╚~ūāōQ╝t═Ō(FTIR)╣ŌūVė╔Ą┬ć°▓╝¶ö┐╦╣½╦ŠĄ─Vertex 70╝t═Ō╣ŌūVāx½@Ą├, ūVłD▓╔╝»ĘČć·×ķ4000~400 cm-1, Ęų▒µ┬╩×ķ4 cm-1.
ĪĪĪĪ2.6 ┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zĄ─╬³ĖĮīŹ“×
ĪĪĪĪ£╩┤_ĘQ╚Ī0.02 g SAPFe║═DAPFeĘųäe╝ė╚ļĄĮ40 mLĄ─śėŲĘŲ┐ųą, į┘╝ė╚ļ20 mLę╗Č©ØŌČ╚Ą─┴“╦ßŃ~╚▄ę║.īóśėŲĘŲ┐ų├ė┌║Ń£žš±╩ÄŽõā╚š±╩Ä╬³ĖĮę╗Č©Ģrķg(25 Īµ, 150 rĪżmin-1).ūŅ║¾, ╚▄ę║═©▀^0.45 ”╠m×V─ż, ė├ļŖĖą±Ņ║ŽĄ╚ļxūė¾w░l╔õ╣ŌūVāx(ICP-OES, ŹuĮ“9800)Ęų╬÷×Vę║ųą╩ŻėÓĄ─Cu2+ØŌČ╚.├┐ĮMīŹ“׊∙įOų├1éĆ┐š░ūśė║═3éĆŲĮąąśė, ūŅ║¾ŲĮ║ŌØŌČ╚╚ĪŲĮŠ∙ųĄ.Ė∙ō■╬³ĖĮīŹ“×Ū░║¾Cu2+ØŌČ╚Ą─▓ŅųĄėŗ╦Ń─²─z▓─┴ŽĄ─╬³ĖĮ┴┐, ╣½╩Į╚ńŽ┬Ż║
ĪĪĪĪ╩Įųą, qt×ķ╬³ĖĮtĢrĄ─╬³ĖĮ┴┐(mgĪżg-1); c0ĪóctĘųäe×ķ╬³ĖĮŪ░║¾Cu2+ØŌČ╚(mgĪżL-1); V×ķ╚▄ę║¾wĘe(L); m×ķ─²─z┘|┴┐(g).
ĪĪĪĪ2.6.1 ╬³ĖĮäė┴”īWīŹ“×
ĪĪĪĪ£╩┤_ĘQ╚Ī0.02 g SAPFe╗“DAPFeų├ė┌20 mL Cu2+│§╩╝ØŌČ╚×ķ200 mgĪżL-1Ą─╚▄ę║ųą, 25 ĪµĪó150 rĪżmin-1Ž┬š±╩ÄĘ┤æ¬, į┌▓╗═¼Ģrķg╚Īśė, ĢrķgįOų├×ķ0.5Īó1Īó2Īó3Īó5Īó7Īó9Īó12Īó20Īó24Īó36 h, ╚▄ę║═©▀^0.45 ”╠m×V─ż║¾, Ęų╬÷╩ŻėÓCu2+ØŌČ╚, ėŗ╦Ń╬³ĖĮ䮥─╬³ĖĮ┴┐.▓╔ė├£╩ę╗╝ēäė┴”īWĪó£╩Č■╝ēäė┴”īW║═Ņw┴Żā╚öU╔ó─Żą═▀MąąöM║Ž, Ųõ▒Ē▀_╩ĮĘųäe╚ńŽ┬Ż║
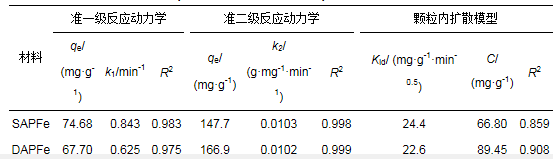
ĪĪĪĪ╩Įųą, k1Īók2║═kidĘųäe╩Ū£╩ę╗╝ēäė┴”īW(min-1)Īó£╩Č■╝ēäė┴”īW(gĪżmg-1min-1)║═Ņw┴Żā╚öU╔ó─Żą═(mgĪżg-1Īżmin-0.5)Ą─╬³ĖĮ╦┘┬╩│ŻöĄ; qe║═qtĘųäe×ķCu2+Ą─ŲĮ║Ō╬³ĖĮ╚▌┴┐(mgĪżg-1)║═į┌tĢrĄ─╬³ĖĮ┴┐(mgĪżg-1).
ĪĪĪĪ2.6.2 ╬³ĖĮĄ╚£žŠĆīŹ“×
ĪĪĪĪ£╩┤_ĘQ╚Ī0.02 g SAPFe╗“DAPFeų├ė┌20 mL Cu2+│§╩╝ØŌČ╚Ęųäe×ķ10Īó20Īó50Īó100Īó150Īó200 mgĪżL-1Ą─╚▄ę║ųą, 25 ĪµĪó150 rĪżmin-1Ž┬š±╩ÄĘ┤æ¬24 h, ╚▄ę║═©▀^0.45 ”╠m×V─ż║¾, Ęų╬÷╩ŻėÓCu2+ØŌČ╚, ėŗ╦Ń╬³ĖĮ䮥─╬³ĖĮ┴┐.╬³ĖĮĄ╚£žŠĆ▓╔ė├LangmuirĪóFreundlichĪóTemkin─Żą═ĘĮ│╠▀MąąöM║Ž, Ųõ▒Ē▀_╩ĮĘųäe╚ńŽ┬Ż║
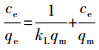
ĪĪĪĪ╩Įųą, KLĪóKFĪóBT║═KTĘųäe×ķLangmuirĪóFreundlich║═Temkin─Żą═ųą┼c╬³ĖĮėąĻPĄ─│ŻöĄ; n×ķFreundlich╬³ĖĮųĖöĄ, ┼c╬³ĖĮ䮥─ąį┘|ėąĻP.
ĪĪĪĪ2.6.3 pHųĄī”┤┼ąį╦«─²─zĄ─ė░Ēæ
ĪĪĪĪ£╩┤_ĘQ╚Ī0.02 g SAPFe╗“DAPFeų├ė┌20 mL Cu2+│§╩╝ØŌČ╚×ķ120 mgĪżL-1Ą─╚▄ę║ųą, š{╣ØCu2+╚▄ę║Ą─pHųĄ×ķ2~6, ▓óįOų├╬┤╠Ē╝ė╦«─²─zĄ─┐š░ūī”ššīŹ“×┐╝▓ņCu2+Ą─│┴ĄĒ¼FŽ¾.į┌25 ĪµĪó150 rĪżmin-1Ž┬š±╩ÄĘ┤æ¬, ═©▀^0.45 ”╠m×V─ż║¾, Ęų╬÷╩ŻėÓCu2+ØŌČ╚, ėŗ╦Ń╬³ĖĮ䮥─╬³ĖĮ┴┐.
ĪĪĪĪ3 ĮY╣¹┼cĘų╬÷(Results and analysis)3.1 ┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zĄ─▒Ēš„3.1.1 ą╬├▓╠žš„
ĪĪĪĪłD 1a╩ŪSAPFe║═DAPFeĄ─īŹ╬’ššŲ¼, ┐╔ęįė^▓ņĄĮSAPFe▒Ē├µ╣Ō╗¼, DAPFe▒Ē├µ┤ų▓┌.SAPFeĮø▀^Ę┤Å═└õā÷ĮŌā÷Ą─╬’└ĒĮ╗┬ōĘĮ╩Į╝┤┐╔Ą├ĄĮŠ▀ėą╗ź┤®ŠWĮjĮYśŗĄ─DAPFe, └õā÷ĢrŠ█ęꎮ┤╝ā╚▓┐Ą─╦«ęį▒∙Š¦Ą─ą╬æB┤µį┌ė┌ĘŪŠ¦ģ^ā╚, Įø▀^ĮŌā÷║¾, ā╚▓┐▒∙Š¦╚┌╗»Č°ą╬│╔╗ź┤®Ą─┐ū(Mohammadi et al., 2014).łD 1b╩Ū┤┼ąį╝{├ūFe3O4Ą─SEMłDŽ±, ×ķ┤¾ąĪŠ∙ę╗Ą─╬óŪ“.łD 1cĪó1dĘųäe×ķSAPFeĘ┼┤¾50║═1000▒ČĄ─SEMłDŽ±, Š▀ėąŅÉ╦ŲĘõĖC├║Ą─ČÓ┐ūŠWĀŅĮYśŗ, ā╚▓┐ĮYśŗĘ┼┤¾║¾┐╔┐┤│÷─²─zęčĮø│╔╣”Š∙ä“Ąžžō▌d┴╦┤┼ąį╝{├ūFe3O4, į÷╝ė┴╦ā╚▓┐Ą─▒╚▒Ē├µĘe.╚²ŠSČÓ┐ūŠWĮjĮYśŗųąŽÓ╗źĮ╗▓µ▀B═©Ą─┐ūŽČ┐╔ęį╩╣▒╗╬³ĖĮĄ─╚▄┘|ĘųūėöU╔ó═©▀^, ėą└¹ė┌┤┼ąį╦«─²─zį┌╦«╠Ä└ĒŅIė“Ą─æ¬ė├.łD 1eĪó1fĘųäe×ķDAPFeĘ┼┤¾50║═1000▒ČĄ─SEMłDŽ±, ░l¼FDAPFeĄ─ŠWĮjĮYśŗūāĄ├ų┬├▄, ▀@┐╔─▄╩Ūė╔ė┌Š█ęꎮ┤╝Įø▀^└õā÷ĮŌā÷裣hĄ─╬’└ĒĮ╗┬ō╩╣─²─zŪ“ā╚▓┐╬’└Ē└pĮYū„ė├╝ėÅŖ, ─²─zŪ“Ą─Į╗┬ō│╠Č╚╝ė┤¾, ą╬│╔┴╦Ė³├▄╝»Ą─ŠWĮjĮYśŗ.
ĪĪĪĪłD 1

ĪĪĪĪłD 1╦«─²─z╝░┤┼ąį╝{├ūFe3O4Ą─▒Ēė^ą╬├▓(a.┤┼ąį╦«─²─zŪ“Ą─╣ŌīWššŲ¼, b.┤┼ąį╝{├ūFe3O4, cĪód.SAPFe, eĪóf.DAPFe)
ĪĪĪĪ3.1.2 ┐ūŽČ║═▒╚▒Ē├µĘeĘų╬÷
ĪĪĪĪęįĄ¬ÜŌ×ķ╬³ĖĮĮķ┘|, į┌77 K║═ŽÓī”ē║┴”10-6~1.0ĘČć·ā╚▀MąąĄ¬ÜŌ╬³ĖĮ├ōĖĮęį£yČ©▒╚▒Ē├µĘe║═┐ūĮYśŗ, łD 2╩ŪSAPFe║═DAPFeĄ─N2╬³ĖĮ├ōĖĮŪ·ŠĆ║═BJH┐ūÅĮĘų▓╝Ū·ŠĆ(▓ÕłD).ĮY╣¹▒Ē├„, SAPFe║═DAPFeĄ─▒╚▒Ē├µĘeĘųäe×ķ64.54║═89.01 m2Īżg-1, ŽÓ▒╚▌^╬┤žō▌d┤┼ąį╝{├ūFe3O4Ą─║ŻįÕ╦ßŌc/Š█ęꎮ┤╝å╬ĪóļpŠWĮjÅ═║Ž╦«─²─z(14.68 m2Īżg-1Īó27.74 m2Īżg-1)▒╚▒Ē├µĘe┤¾┤¾į÷┤¾.┐ūÅĮų„ę¬Ęų▓╝į┌1.2~6.0 nmų«ķg, ┐ū╚▌ĘeĘųäeį┌2.958║═2.924 nm╠Ä▀_ĄĮūŅ┤¾ųĄ.į┌25~60 nmģ^ķg│÷¼F╚§ĘÕ, šf├„▓─┴ŽŠ▀ėąļpĘÕ┐ūÅĮĘų▓╝Ą─╠ž³c.SAPFe║═DAPFeĄ─ŲĮŠ∙┐ūÅĮ┤¾ąĪĘųäe×ķ3.797║═3.443 nm, ┐ū╚▌¾wĘe×ķ0.061║═0.034 cm3Īżg-1.
ĪĪĪĪłD 2
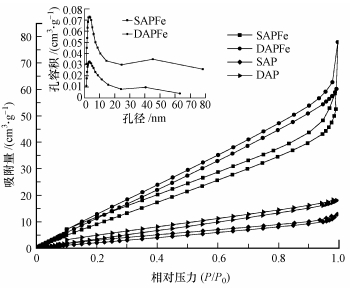
ĪĪĪĪłD 2 SAPFeĪóDAPFeĪóSAP║═DAPĄ─Ą¬ÜŌ╬³ĖĮ├ōĖĮŪ·ŠĆ
ĪĪĪĪ3.1.3 ║¼╦«┬╩Ęų╬÷
ĪĪĪĪłD 3×ķ╝{├ūFe3O4žō▌d┴┐ī”SAPFe║═DAPFeŲĮ║Ō║¼╦«┬╩Ą─ė░ĒæĮY╣¹, Ųõųą, R×ķFe3O4┼c║ŻįÕ╦ßŌcĪóŠ█ęꎮ┤╝┐é┴┐Ą─┘|┴┐▒╚.ė╔łD┐╔ų¬, SAPFeĄ─ŲĮ║Ō║¼╦«┬╩┤¾ė┌DAPFe; ļSų°Fe3O4žō▌d┴┐Ą─į÷ČÓ, ā╔ĘN─²─z▓─┴ŽĄ─ŲĮ║Ō║¼╦«┬╩ĮĄĄ═, ŲõųąŻ¼Fe3O4žō▌d┴┐ūā╗»ī”DAPFeŲĮ║Ō║¼╦«┬╩Ą─ė░ĒæĖ³┤¾.Ęų╬÷┐╔ų¬, ė╔å╬ŠWĮjĄĮļpŠWĮj─²─z, ═©▀^└õā÷ĮŌā÷裣h, ╩╣Š█ęꎮ┤╝░l╔·┴╦Į╗┬ōĘ┤æ¬, ▓╗āHūĶĄK┴╦╦«ī”Ė▀Ęųūėµ£Ą─╚▄ĮŌ, Č°Ūęą╬│╔┴╦Ė³ČÓĄ─Į╗┬ō³c, į÷┤¾┴╦Į╗┬ō│╠Č╚, ╩╣─²─z╩š┐s│╠Č╚į÷┤¾, ą╬│╔Ė³╝ėų┬├▄ĮYśŗ, ę“┤╦, ─²─zĄ─ŲĮ║Ō║¼╦«┬╩ėą╦∙Ž┬ĮĄ(ÓŹĄż, 2014).ļSų°╝{├ūFe3O4žō▌d┴┐Ą─į÷╝ė, ╝{├ū┴ŻūėĄ─╠Ē╝ėš╝ō■┴╦įŁüĒ╦«Ęųūė╦∙š╝ėąĄ─┐šķg, ╩╣─²─zĖ³╝ėų┬├▄, ═¼ĢrūĶĄK┴╦╦«ĘųūėŽ“─²─zā╚▓┐Ą─öU╔ó, ę“┤╦, ŲĮ║Ō║¼╦«┬╩ę▓ļSų«ĮĄĄ═.║¼╦«┬╩Ą═Ą─╦«─²─zŠ▀ėąĖ³║├Ą─ĘĆČ©ąį║═ÖCąĄąį─▄, Ą½Ųõ╚▄├øąį─▄ĮĄĄ═(Spinks et al., 2006).
ĪĪĪĪłD 3
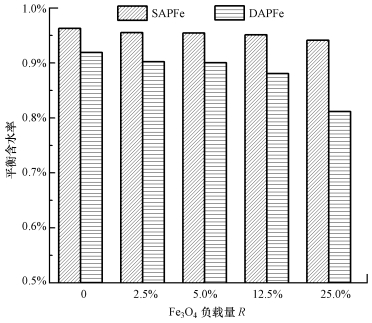
ĪĪĪĪłD 3╝{├ūFe3O4Ą─žō▌dī”SAPFe║═DAPFeŲĮ║Ō║¼╦«┬╩Ą─ė░Ēæ
ĪĪĪĪłD 4×ķ═Ō╝ė┤┼ł÷ū„ė├Ž┬žō▌d0Īó2.5%Īó5%Īó12.5%Īó25%╝{├ūFe3O4Ą─SAPFe║═DAPFeĄ─┤┼ąįą¦╣¹łD, Ųõųą, ╝{├ūFe3O4š╝║ŻįÕ╦ßŌc║═Š█ęꎮ┤╝┐é┴┐12.5%║═25%Ą──²─zŪ“Š∙▒Ē¼F│÷┴╦┴╝║├Ą─┤┼ąį─▄.ĮY║Ž║¼╦«┬╩ĮY╣¹┤_Č©╝{├ū┤┼ąįFe3O4Ą─▀mę╦žō▌d┴┐×ķ12.5%, ▓óū„×ķ║¾└m╬³ĖĮįć“×ųą─²─z▓─┴Žžō▌d┤┼ąį╝{├ūFe3O4Ą─ė├┴┐.
ĪĪĪĪłD 4

ĪĪĪĪłD 4╝{├ūFe3O4Ą─žō▌dī”SAPFe║═DAPFe┤┼ąį─▄Ą─ė░Ēæ(a.¤o═Ō╝ė┤┼ł÷, b.╝ė═Ō╝ė┤┼ł÷)
ĪĪĪĪ3.2 ┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zī”Cu2+Ą─╬³ĖĮąį─▄3.2.1 ╬³ĖĮäė┴”īW
ĪĪĪĪ×ķ┴╦蹊┐SAPFe║═DAPFeī”Cu2+Ą─╬³ĖĮ╦┘┬╩, ī”Ųõ▀Mąą╬³ĖĮäė┴”īWįć“×, ╬³ĖĮäė┴”īW─Żą═öM║ŽĮY╣¹╚ńłD 5a╦∙╩Š.5 hā╚SAPFe║═DAPFeī”Cu2+Ą─╬³ĖĮ┴┐ĘųäečĖ╦┘į÷╝ėĄĮ130.8║═144.2 mgĪżg-1; SAPFeį┌6 hęį║¾╗∙▒Š▀_ĄĮ╬³ĖĮŲĮ║Ō, DAPFeį┌9 h║¾▀_ĄĮ╬³ĖĮŲĮ║Ō; ūŅĮK, SAPFe║═DAPFeī”200 mgĪżL-1 Cu2+Ą─ŲĮ║Ō╬³ĖĮ┴┐Ęųäe×ķ141.8║═162.5 mgĪżg-1; SAPFeī”Cu2+Ą─╬³ĖĮ╦┘┬╩┤¾ė┌DAPFe.DAPFe▒╚▒Ē├µĘe▌^┤¾, ┐ūĮYśŗŽÓī”ų┬├▄, ┐ūÅĮ▌^ąĪ, ę“┤╦, ╬³ĖĮ╦┘┬╩▌^┬².
ĪĪĪĪłD 5
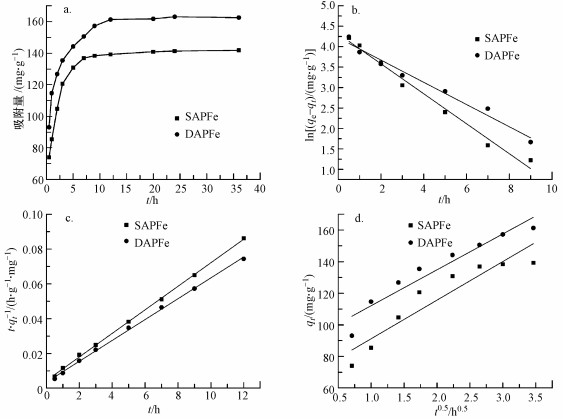
ĪĪĪĪłD 5 SAPFe║═DAPFeī”Cu2+Ą─╬³ĖĮäė┴”īW─Żą═ŠĆąįöM║Ž(a.╬³ĖĮ┴┐ļSĢrķgĄ─ūā╗», b.£╩ę╗╝ēäė┴”īWöM║ŽĮY╣¹, c.£╩Č■╝ēäė┴”īWöM║ŽĮY╣¹, d.Ņw┴Żā╚öU╔ó─Żą═öM║ŽĮY╣¹)
ĪĪĪĪSAPFe║═DAPFeī”Cu2+Ą─╬³ĖĮäė┴”īW─Żą═╚ńłD 5bĪó5cĪó5d╦∙╩Š, öM║ŽģóöĄ╚ń▒Ē 1╦∙╩Š.£╩ę╗╝ēäė┴”īW─Żą═ų╗Ę┤ė│╬³ĖĮ╦┘┬╩┼cę╗ĘNĘ┤æ¬╬’ØŌČ╚Ą─ĻPŽĄ, Š▀ėąŠųŽ▐ąį, Č°£╩Č■╝ēäė┴”īW─Żą═░³║¼═Ō▓┐ę║─żöU╔óĪóŅw┴Żā╚▓┐öU╔ó║═▒Ē├µ╬³ĖĮĄ╚▀^│╠, ─▄ē“Ė³║├ĄžĘ┤ė│╬³ĖĮ▀^│╠║═╬³ĖĮÖC└Ē(ĘĮČžĄ╚, 2016).Ęų╬÷3éĆ─Żą═öM║ŽĄ─┐╔øQŽĄöĄ, ░l¼FSAPFe║═DAPFeĄ─£╩ę╗╝ēäė┴”īW─Żą═Ą─R2Ęųäe×ķ0.90║═0.97, £╩Č■╝ēäė┴”īW─Żą═Ą─R2Š∙┤¾ė┌0.99, öM║ŽČ╚Ė▀ė┌ę╗╝ēäė┴”īW─Żą═, Ųõėŗ╦Ń│÷Ą─ŲĮ║Ō╬³ĖĮ╚▌┴┐qeę▓Ė³ĮėĮ³īŹ“×öĄō■, ╝┤╗»īWĘ┤æ¬╩Ūų„ꬥ─┐ž╦┘▓Į¾E.ė╔Ņw┴Żā╚öU╔ó─Żą═öM║ŽĄ─ĮY╣¹┐╔ų¬, SAPFe║═DAPFeī”Cu2+Ą─╬³ĖĮĘų×ķ2éĆļAČ╬Ż║Ą┌ę╗éĆŠĆąįļAČ╬(t < 15 h)×ķCu2+į┌─²─z▒Ē├µĄ─öU╔ó▀^│╠, Ą┌2éĆļAČ╬(t>15 h)×ķā╚▓┐öU╔ó.ļSų°╬³ĖĮĄ─▀Mąą, SAPFe║═DAPFe▒Ē├µ┐╔└¹ė├Ą─╬³ĖĮ╬╗³cųØu£p╔┘, ╬³ĖĮūŅĮK▀_ĄĮŲĮ║Ō(Li et al., 2013).qt┼ct-0.5öM║Ž×ķŠĆąįĻPŽĄ, Ą½▓╗▀^įŁ³c, šf├„Ņw┴Żā╚öU╔ó╩Ūå╬ĪóļpŠWĮj┤┼ąį╦«─²─zī”Cu2+Ą─ų„ę¬┐ž╦┘ę“╦ž, Ą½▓╗╩Ū╬©ę╗Ą─┐ž╦┘▓Į¾E.
ĪĪĪĪ
ĪĪĪĪ3.2.2 ╬³ĖĮĄ╚£žŠĆ
ĪĪĪĪSAPFe║═DAPFeī”Cu2+Ą─Ą╚£ž╬³ĖĮĘųäeė├LangmuirĪóFreundlich║═Temkin─Żą═▀MąąöM║Ž, ĮY╣¹╚ńłD 6╦∙╩Š, öM║ŽģóöĄ╚ń▒Ē 2╦∙╩Š.ĮY╣¹▒Ē├„, Langmuir║═FreundlichĘĮ│╠Ą├ĄĮ┴╦▌^║├Ą─┐╔øQŽĄöĄ, Temkin─Żą═▌^▓Ņ.Ųõųą, Langmuir╬³ĖĮĄ╚£ž─Żą═×ķå╬Ęųūėīė╬³ĖĮ(īOĄ┬ÄøĄ╚, 2016), ┐╔ęįĖ³║├Ąž├Ķ╩÷Cu2+į┌SAPFe║═DAPFe╔ŽĄ─╬³ĖĮąą×ķ.┤╦═Ō, ═©▀^öM║ŽģóöĄėŗ╦Ń│÷SAPFe║═DAPFeĄ─RLŠ∙Įķė┌0~1, ▒Ē├„ęūė┌╬³ĖĮĄ─▀Mąą.Freundlich─Żą═öM║ŽĄ├ĄĮĄ─nŠ∙┤¾ė┌1, šf├„å╬ĪóļpŠWĮj─²─zŠ∙ī”Cu2+ėą▌^║├Ą─╬³ĖĮą¦╣¹(Kilic et al., 2011).ė╔Langmuir─Żą═┐╔Ą├│÷SAPFe║═DAPFeĄ─’¢║═╬³ĖĮ┴┐Ęųäe×ķ173.01║═207.01 mgĪżg-1, ļpŠWĮj─²─z▌^å╬ŠWĮj─²─zĄ─╬³ĖĮą¦╣¹ėą├„’@╠ßĖ▀.Š▀¾w┬ōŽĄ╬█╦«īÜ╗“ģóęŖhttp://www.jianfeilema.cnĖ³ČÓŽÓĻP╝╝ąg╬─Ön
ĪĪĪĪłD 6
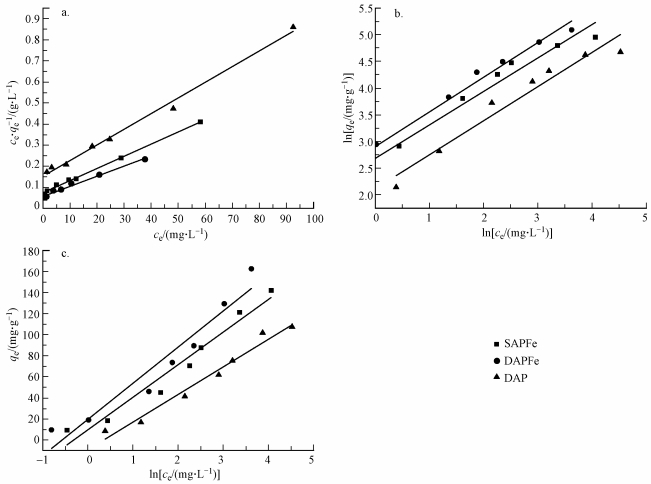
ĪĪĪĪłD 6 SAPFeĪóDAPFe║═DAPī”Cu2+Ą─╬³ĖĮĄ╚£žŠĆ(a. Langmuir, b.Frendlich, c.Tempkin)
ĪĪĪĪ3.3 pHųĄī”┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─z╬³ĖĮĄ─ė░Ēæ
ĪĪĪĪį┌pH×ķ2~6ĘČć·ā╚蹊┐┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zĄ─╬³ĖĮąį─▄, ĮY╣¹╚ńłD 7╦∙╩Š.ļSų°pHĄ─į÷╝ė, ╬³ĖĮ╚▌┴┐į÷┤¾.į┌Ą═pHųĄĢr, ╚▄ę║ųąĄ─H+ęųųŲ┴╦¶╚╗∙║═┴u╗∙Ą─├ō┘|ūėū„ė├, “³║Žū„ė├£p╚§, SAPFe║═DAPFeī”Cu2+Ą─╬³ĖĮ╚▌┴┐Ą═; «öpHį÷┤¾Ģr, H+ØŌČ╚ĮĄĄ═, ¶╚╗∙║═┴u╗∙Ė³╚▌ęū░l╔·├ō┘|ūėū„ė├, ę“Č°┤┘▀M┴╦╬³ĖĮ.┴Ē═Ō, ┐š░ūī”ššīŹ“×ųą░l¼FpHį┌2~5ų«ķg¤o├„’@Ą─Cu2+│┴ĄĒ¼FŽ¾░l╔·, Č°į┌pH=6Ģr, ėą╬ó╚§Ą─│┴ĄĒ¼FŽ¾.ę“┤╦, į┌pH×ķ2~5ų«ķgų„ę¬×ķ▓─┴Žī”Cu2+Ą─╬³ĖĮ, Č°į┌pH=6Ģr, │┴ĄĒū„ė├ī”╬³ĖĮę▓┐╔─▄ėąę╗Č©Ą─žĢ½I.
ĪĪĪĪłD 7
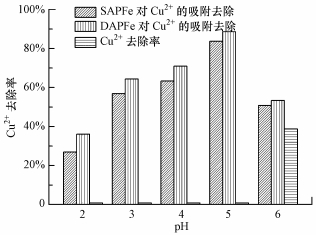
ĪĪĪĪłD 7 pHųĄī”SAPFe║═DAPFe╬³ĖĮCu2+Ą─ė░Ēæ
ĪĪĪĪ3.4 ┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─z╣┘─▄łFĄ─Ęų╬÷
ĪĪĪĪłD 8Įo│÷┴╦SAPFe║═DAPFe╬³ĖĮŃ~ļxūėŪ░║¾į┌4000~400 cm-1ģ^ķgĄ─╣ŌūVłD.SAPFeūVłDųą3279Īó2921Īó1591Īó1412Īó1024 cm-1╠ÄĘųäe×ķĪ¬OHĪóCĪ¬HĪóĪ¬COOHĪ¬(▓╗ī”ĘQ)ĪóCOOĪ¬(ī”ĘQ)ĪóCĪ¬OĪ¬CĄ─╔ņ┐sš±äė(Chhatri et al., 2011), 563 cm-1╠ÄĄ─╬³╩šĘÕ×ķ┤┼ąį╝{├ūFe3O4Ą─╠žš„╬³╩šĘÕ(ńŖ▀h╝tĄ╚, 2010; Cui et al., 2014).ŽÓ▒╚▌^Č°čį, DAPFe╦«─²─zųąė╔ė┌Š█ęꎮ┤╝Į╗┬ō║¾į┌3279Īó2921 cm-1╠ÄĘÕÅŖ£p╚§├„’@, šf├„Š█ęꎮ┤╝ųąĄ─Ī¬OHģó┼c┴╦Į╗┬ōĘ┤æ¬.Š═DAPFeČ°čį, ╬³ĖĮCu2+║¾, Ī¬OH╔ņ┐sš±äėĄ─╬³╩šĘÕė╔╬³ĖĮŪ░Ą─3280 cm-1ęŲäėĄĮ┴╦3295 cm-1; C=OĄ─ī”ĘQ╔ņ┐sš±äėĘÕė╔╬³ĖĮŪ░Ą─1410 cm-1ęŲäėų┴1408 cm-1; CĪ¬OĪ¬CĄ─╔ņ┐sš±äėĄ─╬³╩šĘÕė╔╬³ĖĮŪ░Ą─1021 cm-1ęŲäėĄĮ┴╦1029 cm-1(Kamoun et al., 2015).╗∙łF╬³╩šĘÕį┌╬³ĖĮ║¾Š∙ėąŽÓæ¬Ą─£p╚§, šf├„╬³ĖĮ▀^│╠ųą┴u╗∙╔ŽŠ▀ėą╣┬ī”ļŖūėĄ─O┼cųžĮī┘ļxūė░l╔·┴╦“³║Žū„ė├, ║ŻįÕ╦ßŌc└’Ą─C=O╝░├čµIųąĄ─Ī¬OĪ¬ę▓ģó┼c┴╦ųžĮī┘Ą─╬³ĖĮ(Kumar et al., 2017).
ĪĪĪĪłD 8

ĪĪĪĪłD 8 SAPFe║═DAPFe╬³ĖĮCu2+Ū░Īó║¾Ą─FTIRłD
ĪĪĪĪ4 ĮYšō(Conclusions)
ĪĪĪĪ1) ęį║ŻįÕ╦ßŌc║═Š█ęꎮ┤╝×ķ╣Ū╝▄žō▌d┤┼ąį╝{├ūFe3O4Ņw┴Ż│╔╣”║Ž│╔┴╦┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zSAPFe║═DAPFe.Æ▀├ĶļŖńRłD│╩¼F│÷ŅÉ╦ŲĘõĖC├║Ą─╚²ŠSČÓ┐ūŠWĀŅĮYśŗ, SAPFe║═DAPFeųąĄ─╚²ŠSČÓ┐ūŠWĮjĮYśŗųąŽÓ╗źĮ╗▓µ▀B═©Ą─┐ūŽČęūė┌╚▄┘|ĘųūėöU╔ó═©▀^, ėą└¹ė┌┤┼ąį╦«─²─z╬³ĖĮ╚ź│²╬█╚Š╬’.
ĪĪĪĪ2) DAPFeė╔ė┌Į╗┬ō│╠Č╚┤¾, ║¼╦«┬╩Ą═ė┌SAPFe, Ą½║¼╦«┬╩╚į╚╗─▄▀_ĄĮ80%.SAPFe║═DAPFeĄ─▒╚▒Ē├µĘeĘųäe×ķ64.54 m2Īżg-1║═89.01 m2Īżg-1, ŽÓ▒╚▌^╬┤žō▌d┤┼ąį╝{├ūFe3O4Ą─Å═║Ž╦«─²─z▒╚▒Ē├µĘe┤¾┤¾╠ßĖ▀.┘x┤┼║¾┐╔┐ņ╦┘Ęųļx│÷┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─z.į┌pHųĄ2~5ĘČć·ā╚, ļSų°pHĄ─į÷╝ė, ā╔ĘN▓─┴Žī”Cu2+Ą─╬³ĖĮ╚▌┴┐ųØuį÷┤¾.═©▀^╝t═ŌĘų╬÷░l¼F, ŽÓ▒╚▌^SAPFeČ°čį, DAPFeųąĄ─Š█ęꎮ┤╝ģó╝ė┴╦Į╗┬ōĘ┤æ¬ą╬│╔┴╦Ė³ĘĆČ©Ą─┤┼ąį╦«─²─z.SAPFe║═DAPFeŠ▀ėąžSĖ╗Ą─┼cųžĮī┘«a╔·“³║Žū„ė├Ą─¶╚╗∙║═┴u╗∙╣”─▄ąį╣┘─▄łF.
ĪĪĪĪ3) ┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zī”Cu2+Ą─╬³ĖĮ▀^│╠Ę¹║Ž£╩Č■╝ēäė┴”īW─Żą═, SAPFeī”Cu2+Ą─╬³ĖĮ╦┘┬╩┤¾ė┌DAPFe.╬³ĖĮ▀^│╠─▄ē“║▄║├Ąž▒╗Langmuir╬³ĖĮĄ╚£ž─Żą═├Ķ╩÷, šf├„┤┼ąįĖ▀ĘųūėÅ═║Ž╦«─²─zī”Cu2+Ą─╬³ĖĮ╩Ūå╬Ęųūėīė╬³ĖĮ, DAPFeī”Cu2+Ą─ūŅ┤¾╬³ĖĮ┴┐┐╔▀_207.01 mgĪżg-1, ╬³ĖĮąį─▄ā×ė┌SAPFe(173.01 mgĪżg-1).
ĪĪĪĪŠC╔Ž, ┤┼ąįļpŠWĮj╦«─²─zŪ“(DAPFe)╩Ūę╗ĘNŠG╔½Łh▒Ż¤oČŠĄ─╬³ĖĮä®, įŁ┴ŽüĒį┤ÅVĘ║, Š▀ėą¬Ü╠žĄ─ŠWĮjĮYśŗ║═žSĖ╗Ą─╣┘─▄łF, ╬³ĖĮąį─▄ā×┴╝, ╣╠ę║ĘųļxĘĮ▒Ń, ėąę╗Č©Ą─ķ_░lØō┴”║═æ¬ė├Ū░Š░.(üĒį┤Ż║ŁhŠ│┐ŲīWīWł¾ ū„š▀Ż║┐ūÄr)

