┤┼ąįCeO2-Fe3O4Å═║Ž▓─┴ŽĖ▀ą¦╠Ä└Ē╦«ųą╔ķ╬█╚Š
ųąć°╬█╦«╠Ä└Ē╣ż│╠ŠW ĢrķgŻ║2018-8-15 9:32:59
╬█╦«╠Ä└Ē╝╝ąg | ģRŠ█╚½Ū“Łh▒Ż┴”┴┐Ż¼ĮĄĄ═Ų¾śIų╬╬█│╔▒Š
ĪĪĪĪ1 ę²čį(Introduction)
ĪĪĪĪ╔ķ╩Ūę╗ĘN░ļĮī┘į¬╦ž, ¤o│¶¤o╬Č, Ųš▒ķ┤µį┌ė┌╠ņ╚╗╦«¾w║═╣żśIÅU╦«ųą, ▓óŪę▒╗šJ×ķ╩ŪČŠąįūŅ┤¾Ą─¤oÖC╬█╚Š╬’(Pena et al., 2005).¤oÖCĀŅæBŽ┬Ą─╔ķį¬╦žų„ę¬ęįüå╔ķ╦ßĖ∙║═╔ķ╦ßĖ∙ā╔ĘNą╬╩Į┤µį┌ė┌╦«¾wųą, Ūęüå╔ķ╦ßĖ∙Ą─ČŠąį▒╚╔ķ╦ßĖ∙Ė▀10▒Č(Katsoyiannis et al., 2007).ķLŲ┌▒®┬Čė┌Ė▀╔ķŁhŠ│ųąĢ■ī”╚╦¾w«a╔·ųž┤¾╬Ż║”(Pontius et al., 1994;Guha et al., 2003).╩└Įńąl╔·ĮM┐Ś(WHO)ęÄČ©’ŗė├╦«ųą╔ķĄ─ūŅĖ▀Ž▐Č╚×ķ10 ”╠gĪżL-1(Zhang et al., 2013), ╬ęć°Ą─ĪČ╔·╗Ņ’ŗė├╦«ąl╔·ś╦£╩ĪĘę▓▓╔ė├┴╦▀@ę╗ś╦£╩(Nordstrom et al., 2002).ę“┤╦Ė▀ą¦Ą─│²╔ķ╝╝ąg│╔×ķ┴╦Ė„ć°čąŠ┐╚╦åTĄ─蹊┐¤ß³c.─┐Ū░, ’ŗė├╦«╠Ä└ĒųąĄ─│²╔ķĘĮĘ©ų„ę¬░³└©Ż║╗»īW│┴ĄĒ(Dodd et al., 2006)ĪóļxūėĮ╗ōQ(Wang et al., 2000)Īó─ż▀^×V(Teychene et al., 2013)ĪóļŖ╗ņ─²(Hansen et al., 2006)║═╬³ĖĮĘ©(Sun et al., 2017;Gupta et al., 2010)Ą╚.Ųõųą, ╬³ĖĮĘ©ė╔ė┌║åå╬ęūąąĪó╠Ä└Ēą¦┬╩Ė▀Īó│╔▒ŠĄ═┴«, ęč│╔×ķ«öŪ░ūŅėąæ¬ė├Ū░Š░Ą─’ŗė├╦«│²╔ķĘĮĘ©ų«ę╗(└ŅČ©²łĄ╚, 2007).
ĪĪĪĪĮ³─ĻüĒ, ć°ā╚═Ō蹊┐š▀čąųŲ│÷┴╦╗ŅąįõX(Lin et al., 2001)ĪóĶFč§╗»╬’/Üõč§╗»╬’(Feng et al., 2012)ĪóČ■č§╗»Ōü(Tang et al., 2012)Īóč§╗»µV(Mohan et al., 2007)ĪóČ■č§╗»Ōŗ(Zhang et al., 2005)╝░▀ĆįŁąįĮī┘(Mohan et al., 2007)Ą╚ČÓĘNą┬ą═╝{├ū│²╔ķ╬³ĖĮä®.Ųõųą, ĶF╝░Ųõč§╗»╬’┼c╔ķļxūėŠ▀ėąĖ³ÅŖĄ─ėH║═┴”(Lafferty et al., 2005), ŪęųŲéõĘĮĘ©║åå╬, įŁ▓─┴ŽüĒį┤ÅVĘ║, ¼Fęč▒╗ÅVĘ║蹊┐.╚╗Č°, å╬ę╗ĮMĘ▌ĶFč§╗»╬’┼cAs(ó¾)Ą─ėH║═┴”┤¾┤¾╚§ė┌As(ó§) (Sun et al., 2017;Zhang et al., 2007), ī¦ų┬ŲõļyęįĖ▀ą¦╚ź│²╦«ųąĄ─╚²ār╔ķ, Ž▐ųŲ┴╦ŲõÅVĘ║æ¬ė├.×ķ┤╦, └¹ė├ļpĮMĘ▌╬³ĖĮ▓─┴Žųąā╔ĘNĮMĘ▌─▄«a╔·ģf═¼ą¦æ¬Ą─ā×ä▌, 蹊┐š▀ķ_░l┴╦ę╗ŽĄ┴ąFe-Mn(Shan et al., 2013;Zhao et al., 2012)ĪóFe-Ti(═¶┘ÉŲµĄ╚, 2014)Ą╚ĶF╗∙╝{├ūÅ═║Ž▓─┴Ž.Ųõųą, TiO2ė╔ė┌Ņw┴ŻąĪĪó▒╚▒Ē├µĘe┤¾Īó╣Ō┤▀╗»╗ŅąįĖ▀ĪóįņārĄ═┴«ŪęĖ╗║¼ėH╦«ąįĄ─▒Ē├µ┴u╗∙, ┐╔īóŲõų▒Įėžō▌dė┌┤┼ąį▌d¾w╔Ž(Liu et al., 2014).YuĄ╚(2014)ųŲéõ┴╦”├-Fe2O3-TiO2╝{├ū╬³ĖĮä®, į┌ūŽ═Ō╣ŌŽ┬, Å═║Ž╣Ō┤▀╗»╬³ĖĮ䮫a╔·Ą─┴u╗∙ūįė╔╗∙─▄īóAs(ó¾)č§╗»×ķAs(ó§), ▓óīóŲõ▀Mę╗▓Į╬³ĖĮė┌ĶFč§╗»╬’▒Ē├µ, ▀_ĄĮ│²╔ķĄ──┐Ą─.Ą½ė╔ė┌TiO2┼c”├-Fe2O3Å═║Žč┌╔w┴╦ĶF▓─┴Ž▒Ē├µĄ─╗Ņąį╬³ĖĮ³c╬╗, į┘╝ė╔Ž┴Ēę╗ĮMĘ▌TiO2▓─┴Žī”╔ķĄ─╬³ĖĮ─▄┴”▌^Ą═, ī¦ų┬Ųõš¹¾w│²╔ķą¦─▄▓ó▓╗Ė▀.RaoĄ╚(2015)▓╔ė├╣▓│┴ĄĒĘ©║Ž│╔┴╦Fe-TiļpĮMĘ▌č§╗»╬’, į┌ūŅā׌l╝■Ž┬, Ųõī”╔ķĄ─’¢║═╬³ĖĮ┴┐āHėą31.42 mgĪżg-1.ę“┤╦, žĮ┤²īżŪ¾╝µŠ▀┴╝║├╣Ō┤▀╗»ąį─▄Ą─═¼Ģrī”╦«ųąĄ─╔ķļxūėėą┴╝║├Ą─╬³ĖĮąį─▄Ą─▓─┴Ž, ┼cFe3O4║Ž│╔Å═║Ž▓─┴Ž, ū„×ķ╣Ō┤▀╗»ä®║═╬³ĖĮä®ė├ė┌Ė▀ą¦╠Ä└Ē╦«ųą╔ķ╬█╚Š.
ĪĪĪĪCeO2ū„×ķŽĪ═┴▓─┴Žųąę╗ĘNĖ▀ą¦ĮøØ·Ą─╣Ō┤▀╗»│╔Ęų, ¼Fęčæ¬ė├ė┌╣Ō┤▀╗»ŅIė“(Arul et al., 2013).═¼Ģr, ė╔ė┌ī”╦«ųą╔ķļxūėŠ▀ėą▌^ÅŖĄ─ėH║═┴”, Ōŗ╗∙č§╗»╬’▀Ć▒╗ė├ė┌Ė▀ą¦╬³ĖĮ╚ź│²╦«ųą╔ķļxūė(Gupta et al., 2012).Ķbė┌┤╦, ▒ŠčąŠ┐└¹ė├CeO2░ļī¦¾wĄ─╣Ō┤▀╗»╗Ņąį╝░CeO2║═Fe3O4ī”As(ó§)Ą─ÅŖėH║═┴”, ║Ž│╔ĶFŌŗļpĮMĘ▌┤┼ąį╝{├ū╬³ĖĮ│²╔ķ▓─┴Ž, Å─Č°└¹ė├╣Ō┤▀╗»ū„ė├īóAs(ó¾)č§╗»×ķAs(ó§), ▀MČ°┐╝▓ņ╦«ųąAs(ó¾)Ą─╚ź│²ą¦╣¹╝░╬³ĖĮ│²╔ķĄ─ė░Ēæę“╦ž, į┌┤╦╗∙ĄA╔Ž, 蹊┐┴╦ĶFŌŗÅ═║Ž╬’Ą─╬³ĖĮ╠žąį.
ĪĪĪĪ2 ▓─┴Ž┼cĘĮĘ©(Materials and methods)2.1 īŹ“×▓─┴Ž
ĪĪĪĪüå╔ķ╦ßŌc(Ęų╬÷╝ā)ū„×ķ╚²ār╔ķį┤, ┘Åūį▒▒Š®╗»īWįćä®ÅS.Ž§╦ßŌŗĪó─“╦žĪó┬╚╗»ĶFĪó┬╚╗»üåĶFĪóÜõč§╗»ŌcĪó┴∙üå╝ū╗∙╦─░ĘĄ╚Š∙×ķĘų╬÷╝ā(ć°╦Ä╝»łF╗»īWįćä®ėąŽ▐╣½╦Š).╦∙ėąĄ─ā”éõę║╝░╚▄ę║Š∙▓╔ė├│¼╝ā╦«┼õųŲ, As(ó¾)ā”éõę║Ą─ØŌČ╚×ķ1 gĪżL-1, Ę┼ė┌4 Īµ▒∙Žõųąā”┤µéõė├.
ĪĪĪĪ2.2 Fe-CeÅ═║Ž▓─┴ŽĄ─ųŲéõ
ĪĪĪĪīó4.34 gŽ§╦ßŌŗ║═4.80 g─“╦ž╚▄ė┌150 mL│¼╝ā╦«ųą, Ę┼ų├ė┌90 Īµ║Ń£ž╦«įĪųą, │ų└möć░Ķ12 h.Ę┤æ¬║¾, ╔·│╔░ū╔½│┴ĄĒ, īó░ū╔½æęØßę║└õģsų┴╩ę£ž, ļxą─Ęųļx, ė├│¼╝ā╦«Ž┤£ņ5┤╬, 80 Īµšµ┐šĖ╔į’║¾, ų├ė┌±RĖźĀtųą300 Īµ▒║¤²2 h, čą─ź│╔Ę█─®, ╝┤Ą├Č■č§╗»Ōŗ.
ĪĪĪĪīó1.99 g┬╚╗»üåĶF╚▄ę║║═4.61 g┬╚╗»ĶF╚▄ę║░┤─”Ā¢▒╚1:1.7╝ė╚ļ400 mL│¼╝ā╦«ųą, öć░Ķ╩╣Ųõ═Ļ╚½╚▄ĮŌ.į┘īó1.72 gČ■č§╗»Ōŗ║═1 g┴∙üå╝ū╗∙╦─░Ę╝ė╚ļ╔Ž╩÷╚▄ę║, ▓óīóŲõĘ┼ų├ė┌80 Īµ║Ń£ž╦«įĪ, │ų└m┤┼┴”öć░Ķ═¼ĢrĄ╬╝ė0.1 molĪżL-1Ą─NaOH╚▄ę║, ų▒ų┴╚▄ę║pH =10, └^└möć░Ķ2 h, Ą├ĄĮ║┌╔½│┴ĄĒ, ĻÉ╗»8 h, ▀^×V║¾ė├│¼╝ā╦«Ž┤£ņų┴ųąąį, 80 Īµšµ┐šĖ╔į’, čą─ź║¾Ę┼╚ļ¤o╦«ęę┤╝ųą▒Ż┤µéõė├.
ĪĪĪĪ2.3 ▓─┴Ž▒Ēš„
ĪĪĪĪ▓╔ė├X╔õŠĆč▄╔õāx(Shimadzu XRD-6100, ╚š▒Š)Ęų╬÷Å═║Ž▓─┴ŽĄ─Š¦ą═ĮYśŗ;▓╔ė├Æ▀├ĶļŖūė’@╬óńR(Hitachi S-3500N, ╚š▒Š)▒Ēš„▓─┴ŽĄ─▒Ēė^ą╬├▓;Å═║Ž▓─┴ŽĄ─▒╚▒Ē├µĘeĪó┐ū╚▌║═┐ūÅĮĘų▓╝ė╔▒╚▒Ē├µĘe£yČ©āx(Micromeritics ASAP 2020, ├└ć°)£yČ©;▓╔ė├š±äėśėŲĘ┤┼ÅŖėŗ(JDM-13, ųąć°)ī”Å═║Ž▓─┴Ž▀Mąą┤┼╠žąįĘų╬÷.
ĪĪĪĪ2.4 ╣Ō┤▀╗»/╬³ĖĮīŹ“×
ĪĪĪĪFe-CeÅ═║Ž▓─┴ŽĄ─╣Ō┤▀╗»/╬³ĖĮ│²╔ķīŹ“×į┌│Ż£ž│Żē║Ž┬▀Mąą.ęį300 WūŽ═Ō¤¶ū„×ķ╣Ōį┤, ┼·┴┐įć“ץ─As(ó¾)ØŌČ╚×ķ10 mgĪżL-1, ╬³ĖĮä®ØŌČ╚×ķ200 mgĪżL-1, ļxūėÅŖČ╚(ęįNaNO3ėŗ)×ķ0.01 molĪżL-1, Ę┤æ¬ę║│§╩╝pH×ķ7.0.į┌╩»ėó╣▄ųą╝ė╚ļ50 mL 10 mgĪżL-1Ą─As(ó¾)╚▄ę║║═10 mg Fe-CeÅ═║Ž▓─┴Ž, īó╩»ėó╣▄ų├ė┌╣Ō┤▀╗»Ę┤æ¬Ų„(OCRS-IV TYPE)ųą, į┌ūŽ═Ō╣Ōšš╔õŽ┬, ęį▐D╦┘×ķ150 rĪżmin-1│ų└m┤┼┴”öć░Ķ, ╣ŌĘ┤æ¬30 min║¾, ĻPķ]ūŽ═Ō╣Ōį┤, └^└möć░Ķ690 min, ęį▒ŻūC▀_ĄĮ╬³ĖĮŲĮ║Ō.Č©Ģr╚Īśė, ė├0.45 ”╠m╬ó┐ū×V─żŲ„Ęųļx╚ź│²╦«ųą╝{├ūŅw┴Ż, ė├ļŖĖąĄ╚ļxūė¾w░l╔õ╣ŌūV(ICP-AES)£yČ©×Vę║ųąĄ─┐é╔ķØŌČ╚, ×Vę║ųą╚²ār╔ķ║═╬Õār╔ķĄ─ØŌČ╚▓╔ė├ļxūė╔½ūVĘ©£yČ©.
ĪĪĪĪ3 ĮY╣¹┼cėæšō(Results and discussion)3.1 ▓─┴Ž▒Ēš„3.1.1 ╬óė^ą╬├▓┼cŠ¦¾wĮYśŗ
ĪĪĪĪ└¹ė├Æ▀├ĶļŖūė’@╬óńR(SEM)ė^▓ņ┴╦Fe-CeÅ═║Ž▓─┴ŽĄ─╬óė^ą╬├▓.Å─łD 1ųą┐╔ęį┐┤│÷, Fe-CeÅ═║Ž▓─┴Ž▒Ē├µ┤ų▓┌▓ó│╩¼F┐ūĄ└ĮYśŗ, Å═║Ž▓─┴ŽŅw┴Ż×ķ╝{├ū╝ēĘŪŠ∙ä“┴Żūė, ┴ŻÅĮ╝s×ķ10~30 nm.X╔õŠĆč▄╔õĘų╬÷(XRD)│Żė├ė┌▓─┴ŽĄ─Š¦¾wĮYśŗĘų╬÷, ╚ńłD 2╦∙╩Š, 2”╚į┌10ĪŃ~80ĪŃĘČć·ā╚, │╩¼Fę╗ŽĄ┴ą╠žš„č▄╔õĘÕ.Ųõųąę╗ą®č▄╔õĘÕī┘ė┌Fe3O4 (JCPDs 88-0315)Ą─(220)Īó(311)Īó(400)Īó(422)Īó(511)║═(110)Š¦├µ.į┌33.4ĪŃĪó47.1ĪŃĪó51.7ĪŃĪó59.1ĪŃĪó69.7ĪŃ║═77.1ĪŃ╠ÄĄ─č▄╔õĘÕŠ∙ī┘ė┌CeO2 (JCPDs 65-5923), ▀@šf├„įō║Ž│╔▓─┴Žė╔Fe3O4║═CeO2ĮM│╔, ╝┤×ķCeO2-Fe3O4Å═║Ž▓─┴Ž.
ĪĪĪĪłD 1

ĪĪĪĪłD 1 CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─Æ▀├ĶļŖńRłD
ĪĪĪĪłD 2

ĪĪĪĪłD 2 CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─X╔õŠĆč▄╔õłDūV
ĪĪĪĪ3.1.2 ▓─┴ŽĄ─└Ē╗»ąį┘|
ĪĪĪĪCeO2-Fe3O4Å═║Ž▓─┴ŽĄ─Ą¬ÜŌ╬³ĖĮ├ōĖĮŪ·ŠĆ┼c┐ūÅĮĘų▓╝Ęų╬÷ĮY╣¹╚ńłD 3╦∙╩Š.CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─BET▒╚▒Ē├µĘeĪó┐ū¾wĘe║═ŲĮŠ∙┐ūÅĮĘųäe×ķ174.69 m2Īżg-1Īó0.27 cm3Īżg-1║═15.03 nm.Ė∙ō■ć°ļH╝ā┤Ō┼cæ¬ė├╗»īW┬ō║ŽĢ■(IUPAC)ĘųŅÉ(Ryoo et al., 2001), įōÅ═║Ž╬’Ą─Ą¬ÜŌ╬³ĖĮ├ōĖĮĄ╚£žŠĆī┘ė┌Langmuir ó¶ą═, ×ķĄõą═Ą─Įķ┐ū▓─┴Ž╬³ĖĮŪ·ŠĆ, Ųõ┐ūÅĮĘų▓╝Ū·ŠĆī┘ė┌H3ą═, ▀Mę╗▓Į▒Ē├„įōÅ═║Ž╬’ī┘ė┌Įķ┐ū▓─┴Ž.
ĪĪĪĪłD 3
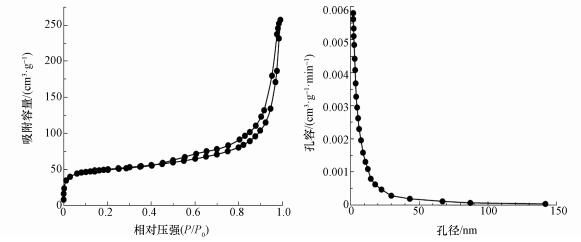
ĪĪĪĪłD 3 CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─▒╚▒Ē├µĘe╝░┐ūÅĮĘų▓╝ (a.CeO2-Fe3O4Ą─Ą¬ÜŌ╬³ĖĮ├ōĖĮŪ·ŠĆ; b.CeO2-Fe3O4Ą─┐ūÅĮĘų▓╝)
ĪĪĪĪ×ķ┴╦╠ĮŠ┐CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─┤┼╠žąį, ╩ę£žŽ┬Å═║Ž▓─┴ŽĄ─┤┼£■╗žŠĆ╚ńłD 4╦∙╩Š.▓─┴ŽĄ─’¢║═┤┼╗»ÅŖČ╚×ķ51.26 emuĪżg-1, ▒Ē├„įō▓─┴ŽŠ▀ėą╚§ĶF┤┼ąį, ┐╔═©▀^═Ō╝ė┤┼ł÷▀MąąĘųļx╗ž╩š.═¼Ģr═©▀^łD 4Ą─║Ļė^┤┼Ęųļxā╚łD┐╔ęį┐┤│÷, CeO2-Fe3O4Å═║Ž▓─┴Žį┌╚▄ę║ųąĄ─Ęų╔óą¦╣¹▌^║├, Ņw┴ŻĘų▓╝Š∙ä“.į┌═Ō╝ė┤┼ł÷Ą─ū„ė├Ž┬, ▓─┴Ž─▄┐ņ╦┘īŹ¼F╣╠ę║Ęųļx, ▀@×ķįō▓─┴ŽĄ─╗ž╩šį┘└¹ė├╠ß╣®Ą─▒ŻšŽ, ę▓Š▀ėąĖ³ÅVĘ║Ą─æ¬ė├Ū░Š░.
ĪĪĪĪłD 4

ĪĪĪĪłD 4 CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─┤┼£■Ū·ŠĆ
ĪĪĪĪ3.2 CeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮ│²As(ó¾)ą¦╣¹čąŠ┐
ĪĪĪĪį┌å╬¬ÜūŽ═Ō╣Ōšš╔õ║═║┌░ĄŚl╝■Ž┬āH╝ė╚ļÅ═║Ž▓─┴ŽĘųäe▀Mąąī”▒╚įć“×, ┐╝▓ņ▓╗═¼īŹ“׌l╝■Ž┬, ¾wŽĄųą┐é╔ķ║═╬Õār╔ķØŌČ╚ļSĘ┤æ¬ĢrķgĄ─ūā╗», ĮY╣¹Ęųäe╚ńłD 5a║═5b╦∙╩Š.āHį┌ūŽ═Ō╣Ōšš╔õŽ┬, ¾wŽĄųą┐é╔ķØŌČ╚ø]ėą░l╔·ūā╗».▀@šf├„į┌ūŽ═Ōšš╔õ, ▓╗╝ė╚ļCeO2-Fe3O4Å═║Ž▓─┴ŽĄ─ŪķørŽ┬, As(ó¾)▓╗─▄▒╗ėąą¦╚ź│².ė╔ė┌╣Ō┤▀╗»ū„ė├āH░l╔·┤▀╗»ä®▒Ē├µ, ę“┤╦┤▀╗»ä®Š▀ėą┴╝║├Ą─╬³ĖĮąį─▄ė╚×ķųžę¬.į┌║┌░ĄŚl╝■Ž┬, ╬³ĖĮ60 min, CeO2-Fe3O4Å═║Ž▓─┴Žī”As(ó¾)Ą─╬³ĖĮ╚ź│²ą¦┬╩╝s×ķ52.62%, ▀@šf├„į┌Č╠Ģrķgā╚CeO2-Fe3O4ī”As(ó¾)Š▀ėą▌^║├Ą─╬³ĖĮą¦╣¹.▒M╣▄As(ó¾)┐╔ęį═©▀^ų▒Įė╬³ĖĮū„ė├╚ź│², Ą½ė╔ė┌╬³ĖĮä®ī”As(ó¾)Ą─ėH║═┴”▀hĄ═ė┌As(ó§)(Sun et al., 2017), ▀_ĄĮ╬³ĖĮŲĮ║ŌĢr, As(ó¾)Ą─╬³ĖĮ╚ź│²ą¦┬╩āH×ķ61.23%.×ķ┴╦▀Mę╗▓Į╠ßĖ▀As(ó¾)Ą─╚ź│²ą¦╣¹, į┌ūŽ═Ōū„ė├Ž┬, ¾wŽĄųą╝ė╚ļCeO2-Fe3O4Å═║Ž▓─┴Ž, ą╬│╔UV/ CeO2-Fe3O4¾wŽĄ, ▓óĘųäeęįUV/ Fe3O4ĪóUV/ CeO2¾wŽĄū„ī”▒╚.į┌CeO2-Fe3O4╣Ō┤▀╗»ū„ė├Ž┬, As(ó¾)Ž╚▒╗č§╗»×ķAs(ó§), Č°║¾▒╗╬³ĖĮė┌CeO2-Fe3O4▒Ē├µ, įō¾wŽĄųąĘ┤æ¬4 h, ╔ķĄ─╚ź│²ą¦╣¹┐╔▀_ĄĮ98%.▀@▒Ē├„į┌ūŽ═Ōšš╔õŽ┬, CeO2-Fe3O4Å═║Ž▓─┴ŽŠ▀ėą┴╝║├Ą─╣Ō┤▀╗»/╬³ĖĮ│²╔ķ─▄┴”.
ĪĪĪĪłD 5
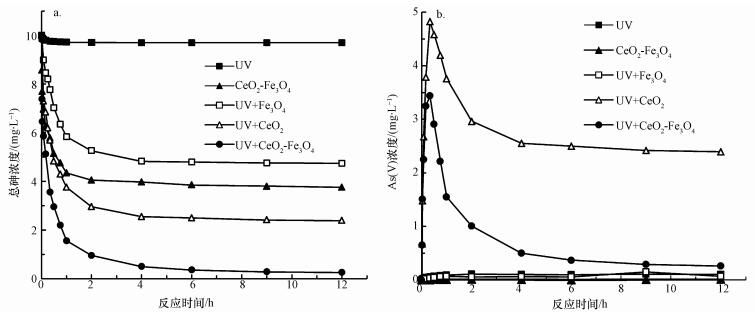
ĪĪĪĪłD 5▓╗═¼Śl╝■Ž┬Ę┤æ¬Ģrķgī”┐é╔ķ(a)║═As(ó§)(b)╚ź│²ą¦╣¹Ą─ė░Ēæ
ĪĪĪĪė╔łD 5b┐╔ų¬, å╬¬ÜūŽ═Ō╣Ō╗“CeO2-Fe3O4┤µį┌Ž┬, ╚▄ę║ųąÄū║§Öz£y▓╗ĄĮAs(ó§)Ą─┤µį┌, ▀@šf├„č§ÜŌ║═ūŽ═Ō▌Ś╔õ▓╗─▄ų▒ĮėīóAs(ó¾)č§╗»×ķAs(ó§).┤╦═Ō, UV/ Fe3O4¾wŽĄųą═¼śėÖz£y▓╗ĄĮAs(ó§)Ą─┤µį┌, ė╔ė┌Fe3O4ø]ėąūŽ═Ō╣Ō┤▀╗»╗Ņąį, į┌ūŽ═Ō╣ŌššŽ┬, UV/ Fe3O4¾wŽĄ▓╗─▄īóAs(ó¾)č§╗»×ķAs(ó§).į┌CeO2╗“CeO2-Fe3O4ūŽ═Ō┤▀╗»ū„ė├Ž┬, ╚▄ę║ųąAs(ó§)Ą─ØŌČ╚Ž╚╔²Ė▀║¾ĮĄĄ═.▀@╩Ūė╔ė┌As(ó§)Ą─ØŌČ╚ė╔As(ó¾)Ą─č§╗»║═As(ó§)Ą─╬³ĖĮā╔▓┐ĘųøQČ©.As(ó¾)č§╗»×ķAs(ó§)╩╣╚▄ę║ųąAs(ó§)Ą─ØŌČ╚╔²Ė▀, Č°As(ó§)╬³ĖĮė┌CeO2-Fe3O4▒Ē├µ╩╣╚▄ę║ųąAs(ó§)Ą─ØŌČ╚ĮĄĄ═.ė╔┤╦┐╔ęŖ, į┌Ę┤æ¬ķ_╩╝Ģr, As(ó¾)Ą─č§╗»ū„ė├š╝ų„ī¦Ąž╬╗.Å─łD 6ųą┐╔ęį┐┤│÷, Ę┤æ¬20 min, ╚▄ę║ųą┐é╔ķØŌČ╚║═As(ó§)Ą─ØŌČ╚ŽÓ═¼, As(ó¾)Ą─ØŌČ╚Äū║§×ķ┴Ń, ╝┤į┌CeO2╗“CeO2-Fe3O4╣Ō┤▀╗»č§╗»ū„ė├Ž┬, As(ó¾)═Ļ╚½▒╗č§╗»×ķĄ═ČŠĄ─As(ó§), ▀@šf├„CeO2║═CeO2-Fe3O4▓─┴ŽŠ∙Š▀ėą▌^║├Ą─ūŽ═Ō╣Ō┤▀╗»╗Ņąį, ▀Mę╗▓ĮūCīŹ┴╦CeO2×ķCeO2-Fe3O4▓─┴ŽųąĄ─╣Ō┤▀╗»╗ŅąįĮMĘ▌.ļS║¾, As(ó§)▒╗╬³ĖĮė┌CeO2-Fe3O4▒Ē├µ, Ą├ĄĮūŅĮK╚ź│², ¾wŽĄųąĄ─┐é╔ķØŌČ╚║═As(ó§)ØŌČ╚ļSų«ĮĄĄ═.
ĪĪĪĪłD 6
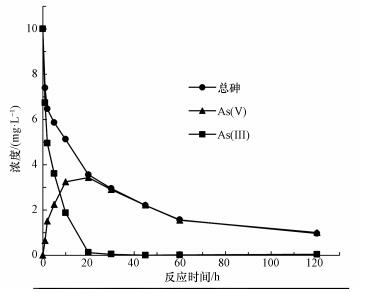
ĪĪĪĪłD 6 CeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮ▀^│╠ųą▓╗═¼ĘNŅÉ╔ķØŌČ╚ūā╗»
ĪĪĪĪ3.3 CeO2-Fe3O4Å═║Ž▓─┴Ž╬³ĖĮ╠žąį蹊┐3.3.1 ╬³ĖĮäė┴”īW
ĪĪĪĪ╬³ĖĮäė┴”īW│Żū„×ķ├Ķ╩÷╬³ĖĮ╦┘┬╩║═╬³ĖĮäėæBŲĮ║ŌĄ─ųĖś╦.Å─łD 7ųą┐╔ęį┐┤│÷, š¹éĆ╬³ĖĮ▀^│╠┐╔ęįĘų×ķā╔éĆļAČ╬, Ą┌ę╗ļAČ╬╩Ū┐ņ╦┘╬³ĖĮ▀^│╠, ╦∙ąĶĢrķg▌^Č╠, Ę┤æ¬2 h╬³ĖĮ╚▌┴┐ęč▀_ĄĮ╬³ĖĮŲĮ║Ō╚▌┴┐Ą─80%ęį╔Ž.▀@╩Ūė╔ė┌CeO2-Fe3O4Å═║Ž▓─┴Žī”╔ķĄ─╬³ĖĮų„ę¬×ķ┐ūĄ└öU╔ó╬³ĖĮ, į┌ūŅ│§ļAČ╬, ╣╠ę║Įń├µĄ─é„┘|öU╔ó▀^│╠▌^┐ņ.Ą┌Č■ļAČ╬╩Ū╬³ĖĮŲĮ║Ō▀^│╠, ęį┴Żūėā╚öU╔ó×ķų„, ╬³ĖĮ╦┘┬╩Ž┬ĮĄ, ╦∙ąĶĢrķg▌^ķL.«ö╔ķØŌČ╚×ķ10 mgĪżL-1Ģr, ▀_ĄĮ╬³ĖĮŲĮ║Ō╦∙ąĶĢrķg×ķ4 h.ę“┤╦, ║¾└mįć“×Ę┤æ¬Įėė|Ģrķg×ķ12 h, ęį┤_▒Ż╬³ĖĮŲĮ║Ō.
ĪĪĪĪłD 7
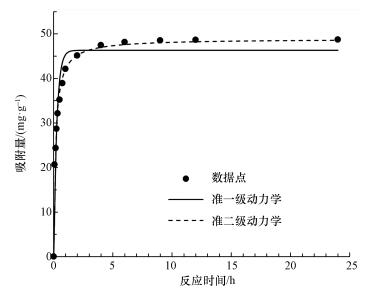
ĪĪĪĪłD 7 CeO2-Fe3O4Å═║Ž╬’╬³ĖĮ│²╔ķĄ─╬³ĖĮäė┴”īW─ŻöM
ĪĪĪĪ╬³ĖĮäė┴”īW─Żą═į┌ę╗Č©│╠Č╚╔Ž─▄ē“ĮŌßī╬³ĖĮÖC└Ē, ┤_Č©╬³ĖĮ╦┘┬╩Ą─┐žųŲ▓Į¾E, Ęųäeė├£╩ę╗╝ēäė┴”īW║═£╩Č■╝ēäė┴”īW─Żą═ī”öĄō■▀Mąąäė┴”īWöM║Ž, ĘĮ│╠╚ń╩Į(1)║═(2)╦∙╩Š.
ĪĪĪĪ╩Įųą, qe×ķ╬³ĖĮŲĮ║ŌĢrĄ─╬³ĖĮ┴┐(mgĪżg-1);qt×ķ╚╬ęŌĢr┐╠Ą─╬³ĖĮ┴┐(mgĪżg-1);t×ķĘ┤æ¬Ģrķg(min);k1×ķ£╩ę╗╝ēäė┴”īW╬³ĖĮ╦┘┬╩│ŻöĄ(min-1);k2×ķ£╩Č■╝ēäė┴”īW╬³ĖĮ╦┘┬╩│ŻöĄ(gĪżmg-1Īżmin-1).
ĪĪĪĪäė┴”īWĘŪŠĆąįöM║ŽĮY╣¹╚ń▒Ē 1╦∙╩Š, ā╔ĘNäė┴”īW─Żą═ī”╬³ĖĮ▀^│╠Ą─öM║ŽČ╚Č╝▀_ĄĮ┴╦0.93ęį╔Ž, Ą½£╩Č■╝ēäė┴”īW─Żą═Ą─öM║ŽČ╚Ė³Ė▀, ┐╔▀_ĄĮ0.99, ▀@šf├„╬³ĖĮ▀^│╠Ė³Ę¹║Ž£╩Č■╝ēäė┴”īW─Żą═.▀@▒Ē├„╬³ĖĮ▀^│╠ų„ę¬ė╔╗»īW╬³ĖĮ×ķų„ī¦, ╬³ĖĮ┘|║═╬³ĖĮä®ų«ķg┤µį┌ļŖūė▐DęŲ▀^│╠(Sun et al., 2017).Š▀¾w┬ōŽĄ╬█╦«īÜ╗“ģóęŖhttp://www.jianfeilema.cnĖ³ČÓŽÓĻP╝╝ąg╬─ÖnĪŻ
ĪĪĪĪ

ĪĪĪĪ3.3.2 ╬³ĖĮĄ╚£žŠĆ
ĪĪĪĪ╬³ĖĮĄ╚£žŠĆ│Żė├ė┌įuār╬³ĖĮ䮥─╬³ĖĮ─▄┴”.▒ŠčąŠ┐▀xė├╚²ār╔ķĄ─│§╩╝ØŌČ╚ĘČć·×ķ1~50 mgĪżL-1, ▀Mąą╣Ō┤▀╗»/╬³ĖĮ│²╔ķīŹ“×, ▓óĘųäe▓╔ė├Langmuir║═Freundlich╬³ĖĮĄ╚£žŠĆī”öĄō■▀MąąöM║Ž, Langmuir║═Freundlich╬³ĖĮĄ╚£ž─Żą═ĘĮ│╠╚ń╩Į(3)║═(4)╦∙╩Š.
ĪĪĪĪ╩Įųą, ce×ķ╬³ĖĮŲĮ║ŌĢr╚▄ę║ųą╔ķļxūėĄ─ØŌČ╚(mgĪżL-1);qe×ķ╬³ĖĮŲĮ║ŌĢrĄ─╬³ĖĮ┴┐(mgĪżg-1);qm×ķ’¢║═╬³ĖĮ┴┐(mgĪżg-1);KL×ķLangmuir│ŻöĄ;n, KF×ķFreundlich│ŻöĄ.
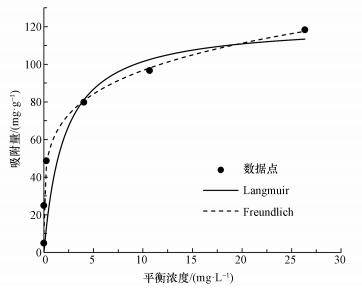
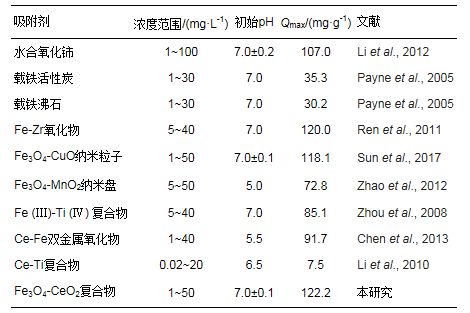
ĪĪĪĪė╔łD 8┐╔ų¬, ļSų°As(ó¾)│§╩╝ØŌČ╚Ą─į÷╝ė, ╬³ĖĮŲĮ║Ō╚▌┴┐ļSų«į÷┤¾.«ö│§╩╝ØŌČ╚į÷┤¾ĄĮę╗Č©│╠Č╚Ģr, ╬³ĖĮĄ╚£žŪ·ŠĆį÷ķL╦┘┬╩ŠÅ┬², ╬³ĖĮ┌ģė┌’¢║═.┤╦═Ō, Ęųäe▓╔ė├Langmuir║═Freundlich╬³ĖĮĄ╚£ž─Żą═▀MąąĘŪŠĆąįöM║Ž, ŽÓĻPģóöĄ╚ń▒Ē 2╦∙╩Š.öM║ŽĮY╣¹▒Ē├„, Langmuir║═Freundlich╬³ĖĮĄ╚£ž─Żą═Ą─öM║ŽĮY╣¹Č╝┴╝║├, öM║ŽČ╚R2Č╝▀_ĄĮ┴╦0.95ęį╔Ž.Ą½▒╚▌^Č°čį, Freundlich─Żą═Ą─öM║ŽČ╚Ė³Ė▀(R2= 0.99).▀@╩Ūė╔ė┌Langmuir╬³ĖĮĄ╚£ž─Żą═╗∙ė┌å╬īė╬³ĖĮ└Ēšō═Ųī¦, Č°Freundlich╬³ĖĮĄ╚£ž─Żą═Ą─└Ēšō╗∙ĄA×ķČÓŽÓ╬³ĖĮ.į┌╣Ō┤▀╗»/╬³ĖĮ▀^│╠ųą, ╚²ār╔ķĄ─╬³ĖĮ▀^│╠░ķļSų°č§╗»▀ĆįŁĘ┤æ¬Ą─░l╔·, ▀@╩Ūę╗ĘNĘŪŠ∙ŽÓ╬³ĖĮ▀^│╠, ╦∙ęį╚²ār╔ķĄ─╬³ĖĮ▀^│╠Ė³▀mė├ė┌Freundlich╬³ĖĮĄ╚£ž─Żą═├Ķ╩÷.į┌Freundlich╬³ĖĮĄ╚£ž╩Įųą, n╩Ūę╗éĆ┼c£žČ╚Ą╚ę“╦žėąĻPĄ─│ŻöĄ, ═©│Żė├üĒįuār╬³ĖĮä®▒Ē├µ╬³ĖĮ³c╬╗Ą─▓╗Š∙ä“ąį║═╬³ĖĮĮY║Ž┴”Ą─┤¾ąĪ, ╦³Ą─ųĄįĮ┤¾▒Ē├„╬³ĖĮä®▒Ē├µįĮ▓╗Š∙ä“, ╬³ĖĮĮY║Ž┴”įĮ┤¾(Shan et al., 2013;Ren et al., 2011).Å─öM║ŽĄ─ĮY╣¹┐╔ęį┐┤│÷n=2.61, ▀Mę╗▓Įšf├„CeO2-Fe3O4╬³ĖĮ䮥─▒Ē├µĄ─▓╗Š∙ä“ąį, ╬³ĖĮĮY║Ž┴”▒╚▌^┤¾, Š▀ėąę╗Č©Ą─╬³ĖĮ─▄┴”.┤╦═Ōė╔Langmuir╬³ĖĮĄ╚£ž─Żą═öM║ŽĮY╣¹Ą├│÷, CeO2-Fe3O4╝{├ū┴ŻūėĄ─╬³ĖĮ’¢║═┴┐×ķ122.19 mgĪżg-1, Ųõ├„’@Ė▀ė┌Ųõ╦¹╬³ĖĮ䮥─’¢║═╬³ĖĮ┴┐(▒Ē 3).┐╔ęŖ, CeO2-Fe3O4╝{├ū┴Żūė─▄ē“Ė▀ą¦Ą─╚ź│²╦«ųąĄ─╔ķ╬█╚Š╬’, Š▀ėąÅVķ¤Ą─æ¬ė├Ū░Š░.
ĪĪĪĪłD 8
ĪĪĪĪłD 8 CeO2-Fe3O4Å═║Ž╬’╬³ĖĮ│²╔ķĄ─╬³ĖĮĄ╚£žŠĆ─ŻöM
ĪĪĪĪ▒Ē 2 ╔ķ╬³ĖĮĄ─╬³ĖĮĄ╚£žŠĆ─Żą═ģóöĄĪĪĪĪ

▒Ē 3 ▓╗═¼╬³ĖĮä®ī”╔ķĄ─ūŅ┤¾’¢║═┴┐▒╚▌^
ĪĪĪĪ3.4 CeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮė░Ēæę“╦ž3.4.1 │§╩╝pHĄ─ė░Ēæ
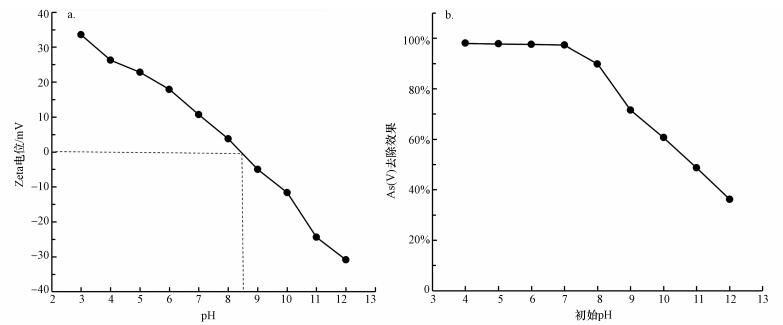
ĪĪĪĪĘųäe▓╔ė├0.1 molĪżL-1 HNO3║═0.1 molĪżL-1 NaOHš{╣Ø│§╩╝pHųĄ, ┐╝▓ņ│§╩╝pHī”CeO2-Fe3O4Å═║Ž╬’╣Ō┤▀╗»/╬³ĖĮ│²╔ķą¦╣¹Ą─ė░Ēæ.Å─łD 9bųą┐╔ęį┐┤│÷, pHį┌4~8ĘČć·ā╚, ╚▄ę║ųą╔ķĄ─╚ź│²┬╩▒Ż│ųį┌98%ū¾ėę, ▀@▒Ē├„į┌ųąąį╗“╦ßąįŚl╝■Ž┬, CeO2-Fe3O4Å═║Ž┴Żūėī”╔ķŠ▀ėą▌^Ė▀Ą─╚ź│²ą¦╣¹.╚╗Č°, pHį┌9~12ĘČć·ā╚, ╔ķĄ─╚ź│²┬╩čĖ╦┘£pąĪų┴▓╗ūŃ35%.ėąčąŠ┐╚╦åT░l¼F╬Õār╔ķĄ─╬³ĖĮ╩▄pHĄ─ė░Ēæ▌^┤¾, ė╚Ųõ╩Ūį┌pH>7Ą─Śl╝■Ž┬, ▓óŪęļSų°pHĄ─į÷┤¾╩▄ė░ĒæĄ─│╠Č╚ę▓įĮüĒįĮ┤¾(Zhang et al., 2007).▀@╩Ūę“×ķ╬Õār╔ķį┌pH < 6.8Ą─Ģr║“į┌╦«ųąęįH2AsO4-Ą─ą╬╩Į┤µį┌, į┌Ė³Ė▀pH╚▄ę║ųąęįHAsO42-Ą─ą╬╩Į┤µį┌.«ö╚▄ę║Ą─pHąĪė┌╬³ĖĮ䮥─Ą╚ļŖ³cĄ─Ģr║“, ╬³ĖĮä®▒Ē├µĄ─┴u╗∙Ģ■┘|ūė╗»ūā│╔OH2+, ▓óį÷ÅŖ╬³ĖĮä®▒Ē├µī”╬Õār╔ķĄ─╬³ĖĮ┴”.ļSų°pHĄ─į÷┤¾, ╬³ĖĮä®▒Ē├µųØuūā×ķĦžōļŖ, ▀@ĘN╬³ĖĮ┴”ę▓Š═įĮüĒįĮ╚§, ī¦ų┬╬³ĖĮ䮥─╬³ĖĮ─▄┴”Ž┬ĮĄ.ė╔łD 9a┐╔ų¬, CeO2-Fe3O4╝{├ū┴ŻūėĄ─Ą╚ļŖ³c×ķ8.5, ╦∙ęį«öpH < 8.5Ģr, ╬³ĖĮä®▒Ē├µÄ¦š²ļŖ║╔, ┤╦Ģr╬Õār╔ķį┌╦«ųą╩ŪęįH2AsO4-║═HAsO42-Ą─ą╬╩Į┤µį┌Ą─, ═©▀^ņoļŖę²┴”ū„ė├┐╔ęį╠ßĖ▀╬³ĖĮä®ī”╬Õār╔ķĄ─╬³ĖĮ─▄┴”, Å─Č°╠ßĖ▀┴╦╔ķĄ─╚ź│²┬╩.į┌ēAąįŚl╝■Ž┬(pH>8.5), ╬³ĖĮä®▒Ē├µÄ¦žōļŖ║╔, Č°╬Õār╔ķūį╔ĒĦėąĖ³ČÓĄ─žōļŖ║╔, ų„ę¬ė╔AsO43-Ą─ą╬╩Į┤µį┌.ė╔ė┌ņoļŖ│Ō┴”Ą─ū„ė├ĮĄĄ═┴╦╬³ĖĮä®ī”╔ķĄ─╬³ĖĮ─▄┴”, ī¦ų┬┴╦╔ķ╚ź│²┬╩Ą─ĮĄĄ═.
ĪĪĪĪłD 9
ĪĪĪĪłD 9 Zeta(a)╝░│§╩╝pHī”CeO2-Fe3O4Å═║Ž╬’╣Ō┤▀╗»/╬³ĖĮ│²╔ķą¦╣¹Ą─ė░Ēæ(b)
ĪĪĪĪ3.4.2 ╣▓┤µļxūėĄ─ė░Ēæ
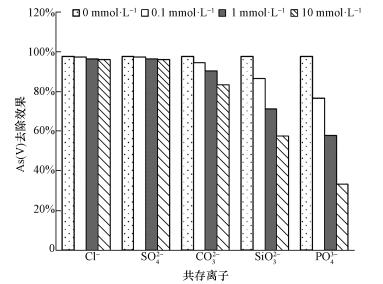
ĪĪĪĪį┌ūį╚╗╦«¾wųą, ╣▓┤µļxūė║═╔ķ╦ßĖ∙ų«ķgą╬│╔ĖéĀÄĻPŽĄ, ĮĄĄ═╬³ĖĮä®ī”╔ķĄ─╚ź│²─▄┴”.ę“┤╦▒ŠčąŠ┐┐╝▓ņ┴╦Cl-ĪóSO42-ĪóCO32-ĪóSiO32-║═PO43-5ĘN│ŻęŖĻÄļxūėī”╔ķ╚ź│²ą¦╣¹Ą─ė░Ēæ, ĮY╣¹╚ńłD 10╦∙╩Š.Cl-║═SO42-Ą─┤µį┌ī”As(ó§)Ą─╬³ĖĮÄū║§ø]ėąė░Ēæ, Č°CO32-ĪóSiO32-║═PO43-Ą─┤µį┌ī”╔ķĄ─╬³ĖĮėąęųųŲū„ė├, ŪęļSų°ĖéĀÄļxūėØŌČ╚Ą─į÷╝ė, ╔ķĄ─╬³ĖĮą¦╣¹’@ų°ĮĄĄ═.═©│ŻŪķørŽ┬, Cl-ų╗─▄║═ĶFĄ─č§╗»╬’╗“Üõč§╗»╬’ą╬│╔═Ōīė┼õ║Ž╬’(Zhang et al., 2007).SO42-║═ĶFč§╗»╬’ų«ķgą╬│╔ā╚īė╗“═Ōīė┼õ║Ž╬’╚ĪøQė┌╚▄ę║pH, į┌pH>6Ģr, SO42-ų„ę¬╩Ū═©▀^═Ō┼õ╬╗ū„ė├╬³ĖĮĄĮ╬³ĖĮä®▒Ē├µ(Shan et al., 2013).ŽÓĘ┤Ąž, AsO43-ät═©▀^┼õ╬╗¾wĮ╗ōQĄ─ÖCųŲ┼c╬³ĖĮä®ų«ķgą╬│╔└╬╣╠Ą─ā╚īė┼õ║Ž╬’(Zhang et al., 2007).ę“┤╦, Cl-║═SO42-Ą─┤µį┌ī”╔ķĄ─╬³ĖĮą¦╣¹ø]ėą├„’@ė░Ēæ.╚╗Č°, ╣▓┤µĄ─SiO32-ĪóPO43-║═ĶFč§╗»╬’╗“Üõč§╗»╬’ų«ķgą╬│╔ā╚īė┼õ║Ž╬’(Zhang et al., 2009), Å─Č°┼cAsO43-ĖéĀÄ╬³ĖĮ³c╬╗, ī¦ų┬╔ķĄ─╚ź│²ą¦╣¹ĮĄĄ═.┤╦═Ō, ┴ū┼c╔ķį¬╦žī┘ė┌═¼ę╗ų„ūÕ, ╗»īWąį┘|ŽÓ╦Ų, ī”╔ķĄ─ĖéĀÄ─▄┴”ūŅÅŖ.ī”ė┌CO32-üĒšf, ╝┤╩╣CO32-║═AsO43-Š∙┐╔ęį┼cĶFĄ─č§╗»╬’╗“Üõč§╗»╬’ų«ķg«a╔·ā╚┼õ╬╗ū„ė├, ą╬│╔ā╚īė┼õ║Ž╬’, Ą½╠╝╦ß¹}Ą─ėH║═┴”ę¬┤¾┤¾Ą═ė┌╔ķ╦ß¹}, ╩╣Ųõī”╔ķĄ─╬³ĖĮė░ĒæŽÓī”▌^ąĪ(Brechb©╣hl et al., 2012).
ĪĪĪĪłD 10
ĪĪĪĪłD 10╣▓┤µļxūėī”CeO2-Fe3O4Å═║Ž╬’╣Ō┤▀╗»/╬³ĖĮ│²╔ķą¦╣¹Ą─ė░Ēæ
ĪĪĪĪ3.5 CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─ųžÅ═└¹ė├ąį
ĪĪĪĪĮøØ·║åå╬Ą─į┘╔·ĘĮĘ©║═┴╝║├Ą─ųžÅ═└¹ė├ąį╩Ū╬³ĖĮ▓─┴Žį┌īŹļHæ¬ė├ųąĄ─ĻPµI.▓╔ė├0.5 molĪżL-1 NaOH║═0.1 molĪżL-1 NaClū„×ķį┘╔·ä®, ═©▀^裣h└¹ė├5┤╬Å═║Ž▓─┴ŽĄ─╣Ō┤▀╗»/╬³ĖĮĪó├ōĖĮą¦┬╩ūā╗», ┐╝▓ņCeO2-Fe3O4Å═║Ž▓─┴ŽĄ─ųžÅ═└¹ė├ąį.ė╔łD 11┐╔ų¬, Įø▀^├┐┤╬裣hīŹ“×║¾, ╔ķĄ─╚ź│²ą¦╣¹Š∙ėąą®įSŽ┬ĮĄ, Ą½ĮĄĄ═┬╩Š∙į┌3%ų«ā╚.裣h└¹ė├5┤╬║¾, ╣Ō┤▀╗»/╬³ĖĮ│²╔ķą¦┬╩╚į┐╔▀_ĄĮ90%ęį╔Ž, į┘╔·├ōĖĮą¦┬╩┐╔ĄĮ▀_84%.┐╔ęŖ, ČÓ┤╬裣h╩╣ė├║¾, ▓─┴ŽĮø▀^į┘╔·╠Ä└Ē╚į─▄▒Ż│ų▌^║├Ą─┤▀╗»/╬³ĖĮ─▄┴”, ▀@šf├„CeO2-Fe3O4Å═║Ž▓─┴ŽŠ▀ėą┴╝║├Ą─ųžÅ═└¹ė├ąį─▄.
ĪĪĪĪłD 11
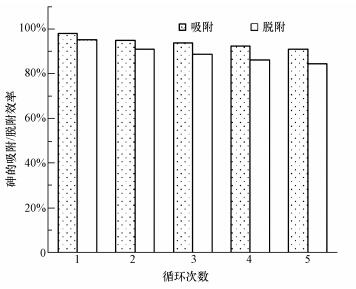
ĪĪĪĪłD 11 CeO2-Fe3O4Å═║Ž╬’Ą─ųžÅ═└¹ė├ąį
ĪĪĪĪ3.6 CeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮ│²As(ó¾)Ą─ÖC└Ē蹊┐
ĪĪĪĪ×ķ┴╦┤_Č©╣Ō┤▀╗»č§╗»▀^│╠ųąĄ─č§╗»╗Ņąį╬’ĘN, į┌UV/ CeO2-Fe3O4¾wŽĄųą╝ė╚ļ▓ČūĮä®▀MąąčąŠ┐, īŹ“×ĮY╣¹╚ńłD 12╦∙╩Š.Ęųäe▀x╚Ī╩ÕČĪ┤╝(t-BuOH), Ą¬ÜŌ(N2)║═╝ū╦ß(FA)ū„×ķ┴u╗∙ūįė╔╗∙(ĪżOH)Īó│¼č§ūįė╔╗∙(Īż O2-)║═╣Ō╔·┐šč©Ą─▓ČūĮä®.į┌╝ū╦ß┤µį┌Śl╝■Ž┬, As(ó¾)Ą─č§╗»ą¦┬╩ø]ėą├„’@ĮĄĄ═, ▀@šf├„į┌As(ó¾)č§╗»▀^│╠ųą, ╣Ō╔·┐šč©▓╗╩Ūč§╗»╗Ņąį╬’ĘN.Č°╝ė╚ļ╩ÕČĪ┤╝╗“═©╚ļĄ¬ÜŌ║¾, As(ó¾)Ą─č§╗»ą¦┬╩├„’@ĮĄĄ═, ▀@šf├„╣Ō╔·ĪżOH║═ĪżO2-╩ŪAs(ó¾)č§╗»Ą─ų„ę¬╗Ņąį╬’ĘN.
ĪĪĪĪłD
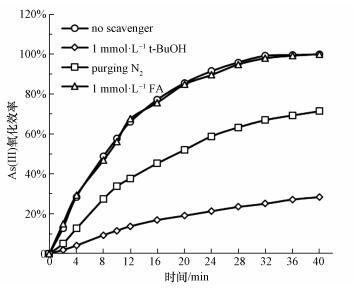
ĪĪĪĪłD 12ūįė╔╗∙▓ČūĮä®ī”As(ó¾)č§╗»ą¦┬╩Ą─ė░Ēæ
ĪĪĪĪCeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮ│²As(ó¾)Ą─ÖC└Ē╚ńłD 13╦∙╩Š.╣Ō┤▀╗»▀^│╠«aĄ─┴u╗∙ūįė╔╗∙║═│¼č§ūįė╔╗∙į┌As(ó¾)Ą─č§╗»▀^│╠ųąŲųžę¬ū„ė├.į┌ūŽ═Ō╣Ōšš╔õŽ┬, ¾wŽĄųąĄ─╦«║═╚▄ĮŌč§į┌CeO2-Fe3O4Å═║Ž▓─┴ŽĄ─╣Ō┤▀╗»ū„ė├Ž┬, Ęųäe«a╔·Š▀ėąÅŖč§╗»ąįĄ─ĪżOH║═ĪżO2-, Š▀¾wĘ┤æ¬╩ĮęŖ╩Į(5)~(7).
ĪĪĪĪłD 13
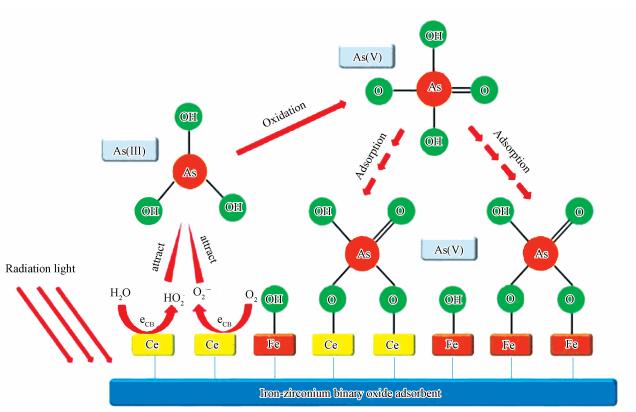
ĪĪĪĪłD 13 CeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮ│²As(ó¾)Ą─ÖC└ĒłD
![]()
ĪĪĪĪė╔ė┌╔·│╔Ą─ūįė╔╗∙Ą─ē█├³║▄Č╠, ╣Ō┤▀╗»č§╗»Ę┤æ¬ČÓ░l╔·į┌╣Ō┤▀╗»ä®▒Ē├µ╗“ĖĮĮ³.╗∙ė┌╔Ž╩÷Ęų╬÷, CeO2-Fe3O4Å═║Ž▓─┴Ž╣Ō┤▀╗»/╬³ĖĮ│²As(ó¾)Ą─▀^│╠ų„ę¬Ęų×ķ3éĆļAČ╬.╩ūŽ╚, ė╔ė┌CeO2-Fe3O4Å═║Ž▓─┴Žī”╔ķŠ▀ėą┴╝║├Ą─╬³ĖĮąį─▄, ╦«ųąAs(ó¾)┐ņ╦┘╬³ĖĮė┌Å═║Ž▓─┴Ž▒Ē├µ.ŠoĮėų°, ╬³ĖĮė┌▓─┴Ž▒Ē├µ║═ĖĮĮ³Ą─As(ó¾)▒╗╣Ō╔·ūįė╔╗∙č§╗»×ķAs(ó§).ė╔ė┌╣Ō┤▀╗»č§╗»▀^│╠Ę┤æ¬čĖ╦┘, į┌┤╦▀^│╠ųąAs(ó¾)Ą─č§╗»╦┘┬╩▀hĖ▀ė┌As(ó§)Ą─╬³ĖĮ╦┘┬╩.ūŅĮK, ╚▄ę║ųąĄ─As(ó¾)▒╗═Ļ╚½č§╗»×ķAs(ó§), As(ó§)▒╗╬³ĖĮė┌CeO2-Fe3O4Å═║Ž▓─┴Ž▒Ē├µĄ├ęį╚ź│².┤╦╣Ō┤▀╗»/╬³ĖĮ│²As(ó¾)ÖC└Ē─▄ē“║▄║├Ą─ĮŌßī╔Ž╩÷īŹ“×ĮY╣¹, ūCīŹ┴╦CeO2-Fe3O4Å═║Ž▓─┴Ž╝µŠ▀┴╝║├Ą─╣Ō┤▀╗»╗Ņąį║═╬³ĖĮąį─▄.
ĪĪĪĪ4 ĮYšō(Conclusions)
ĪĪĪĪ1) ┤┼ąįCeO2-Fe3O4Å═║Ž╬’×ķĮķ┐ū▓─┴Ž, ▒╚▒Ē├µĘeĪóŲĮŠ∙┐ūÅĮĘųäe×ķ174.69 m2Īżg-1║═15.03 nm;▓óŠ▀ėąĘĆČ©Ą─╚§ĶF┤┼ąį, ’¢║═┤┼╗»ÅŖČ╚×ķ51.26 emuĪżg-1.
ĪĪĪĪ2) į┌ūŽ═Ōū„ė├Ž┬, As(ó¾)─▄═Ļ╚½▒╗č§╗»×ķČŠąį▌^Ą═Ą─As(ó§), ═¼ĢrīóAs(ó§)Ė▀ą¦╬³ĖĮė┌CeO2-Fe3O4┴Żūė▒Ē├µ.
ĪĪĪĪ3) į┌ųąąįŚl╝■Ž┬, CeO2-Fe3O4┴Żūėī”As(ó§)Ą─’¢║═╬³ĖĮ┴┐×ķ122.19 mgĪżg-1, Ųõī”As(ó§)Ą─╬³ĖĮ▀^│╠ęį╗»īW╬³ĖĮ×ķų„ī¦.
ĪĪĪĪ4) ╣▓┤µļxūėCl-║═SO42-ī”As(ó§)Ą─╬³ĖĮø]ėą’@ų°ė░Ēæ, Č°CO32-ĪóSiO32-║═PO43-┼cAs(ó§)┤µį┌├„’@Ą─ĖéĀÄ╬³ĖĮ, ╩╣As(ó§)Ą─╬³ĖĮ╚ź│²ą¦╣¹├„’@ĮĄĄ═.
ĪĪĪĪ5) CeO2-Fe3O4Å═║Ž╬³ĖĮä®┐╔┐ņ╦┘īŹ¼F╣╠ę║Ęųļx, ╚▌ęūį┘╔·ŪęųžÅ═└¹ė├ąį▌^║├, Š▀ėąę╗Č©Ą─īŹļHæ¬ė├ārųĄ.(üĒį┤Ż║ŁhŠ│┐ŲīWīWł¾ ū„š▀Ż║īO╠ņę╗)

